Adrenal Cortical Carcinoma
Applied Radiology
Published: December 16, 2025
1 Morehouse School of Medicine, Atlanta, Georgia
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 Department of Radiology, Cincinnati Children’s Hospital, University of Cincinnati College of Medicine, Cincinnati, Ohio
* Corresponding author: Richard B. Towbin (rtowbin@gmail.com)
Abstract
Adrenal cortical carcinoma (ACC) is an aggressive malignancy that has a poor prognosis because of how late they are discovered. Children with ACC often have an underlying genetic disorder. When a tumor is present, they may present with nonspecific or Cushingoid symptoms. CT, MRI, and F-18 fluorodeoxyglucose-PET are used in diagnosis.
Keywords
Retroperitoneal, Neoplasm, Abdominal
Categories
Case Summary
A young child presented with abdominal distension, precocious puberty, weight gain, polyphagia, polydipsia, polyuria, and hypertension. On examination, systolic blood pressure was consistently in the high 100s and occasionally into the 200s. A palpable mass was present in the left upper abdomen. The patient had hirsutism and Tanner stage 2 pubertal development. At laboratory evaluation, the cortisol and dehydroepiandrosterone-sulfate (DHEA-S) levels were elevated. The patient was subsequently diagnosed with Li-Fraumeni syndrome.
Imaging Findings
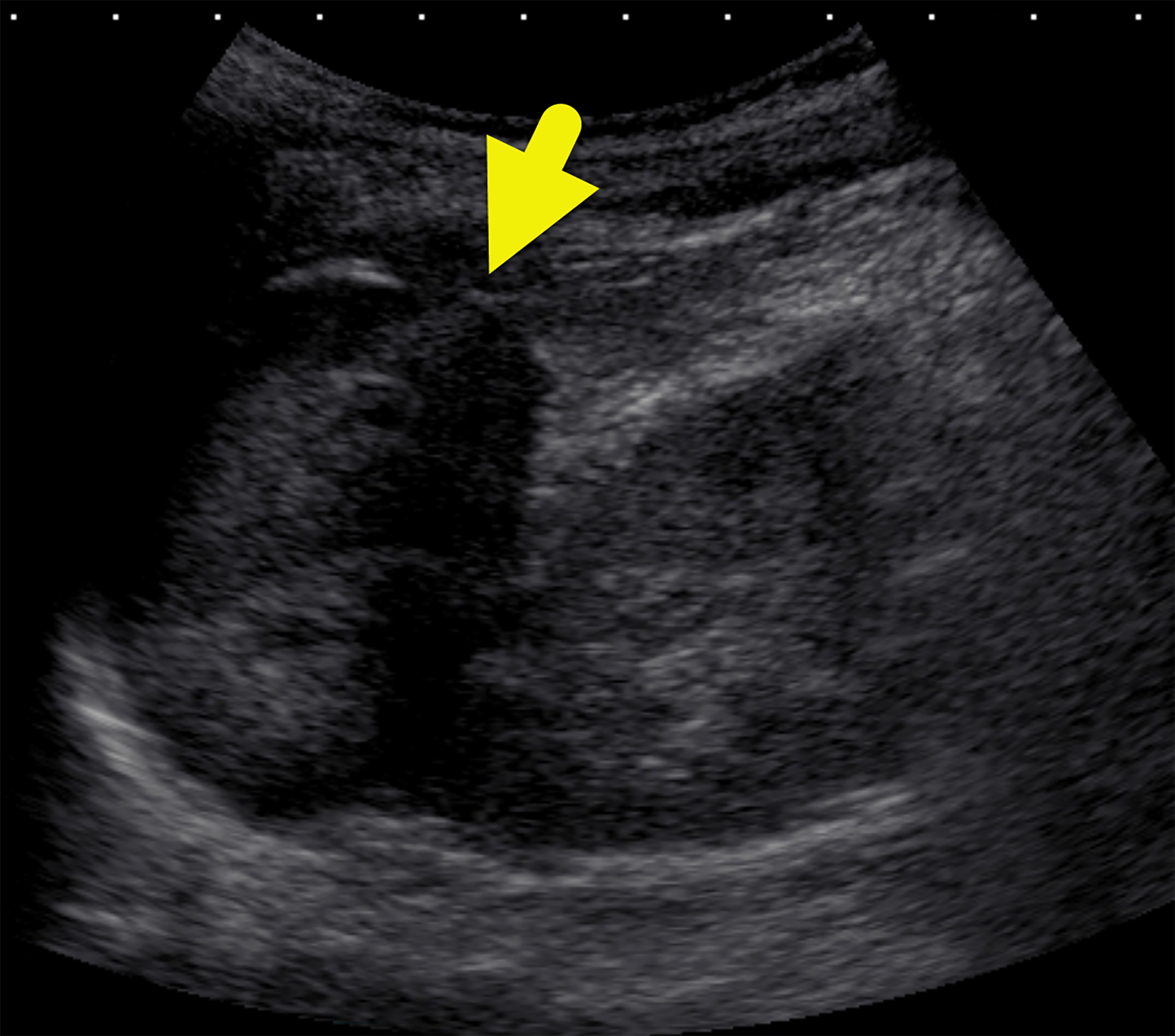
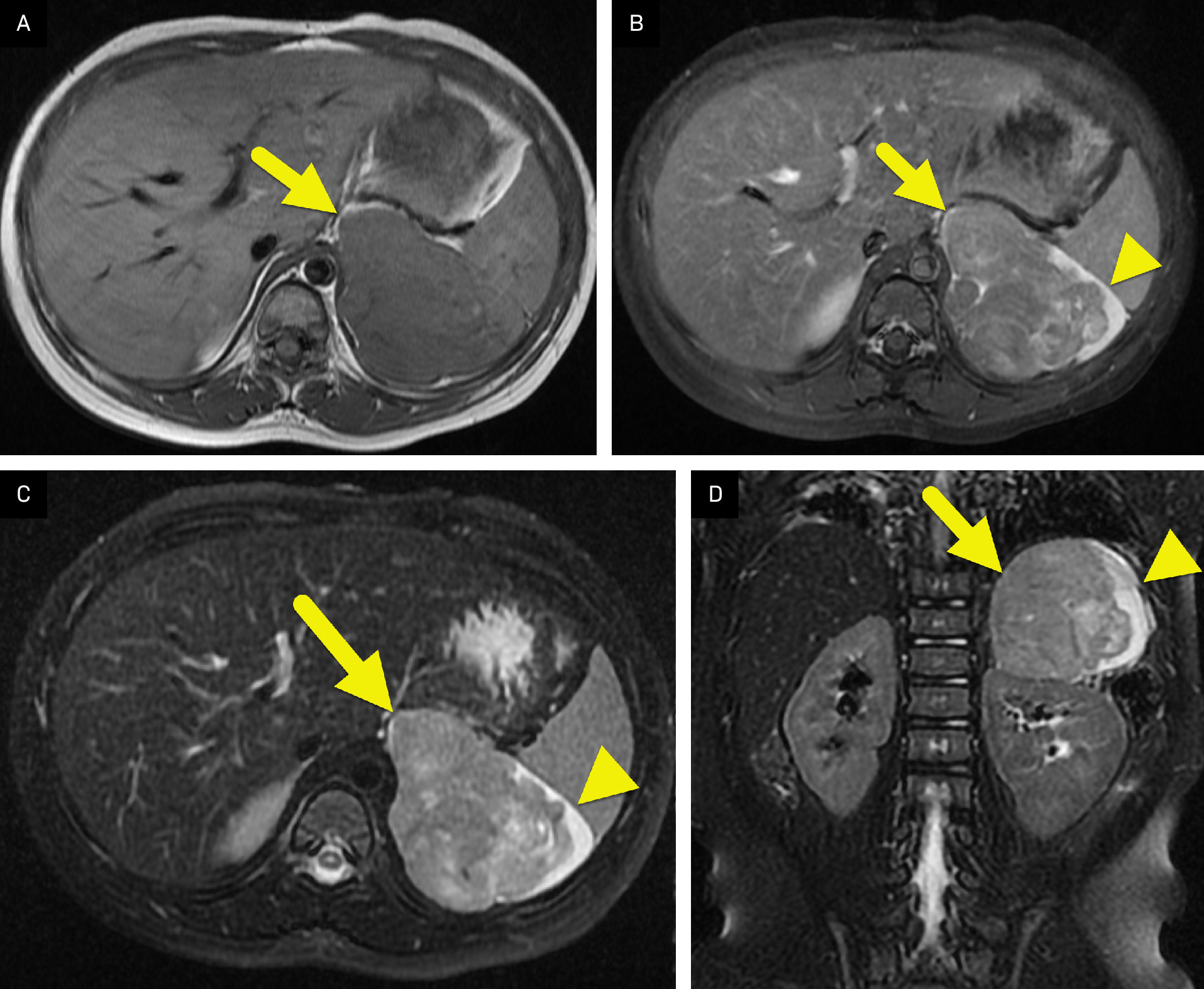
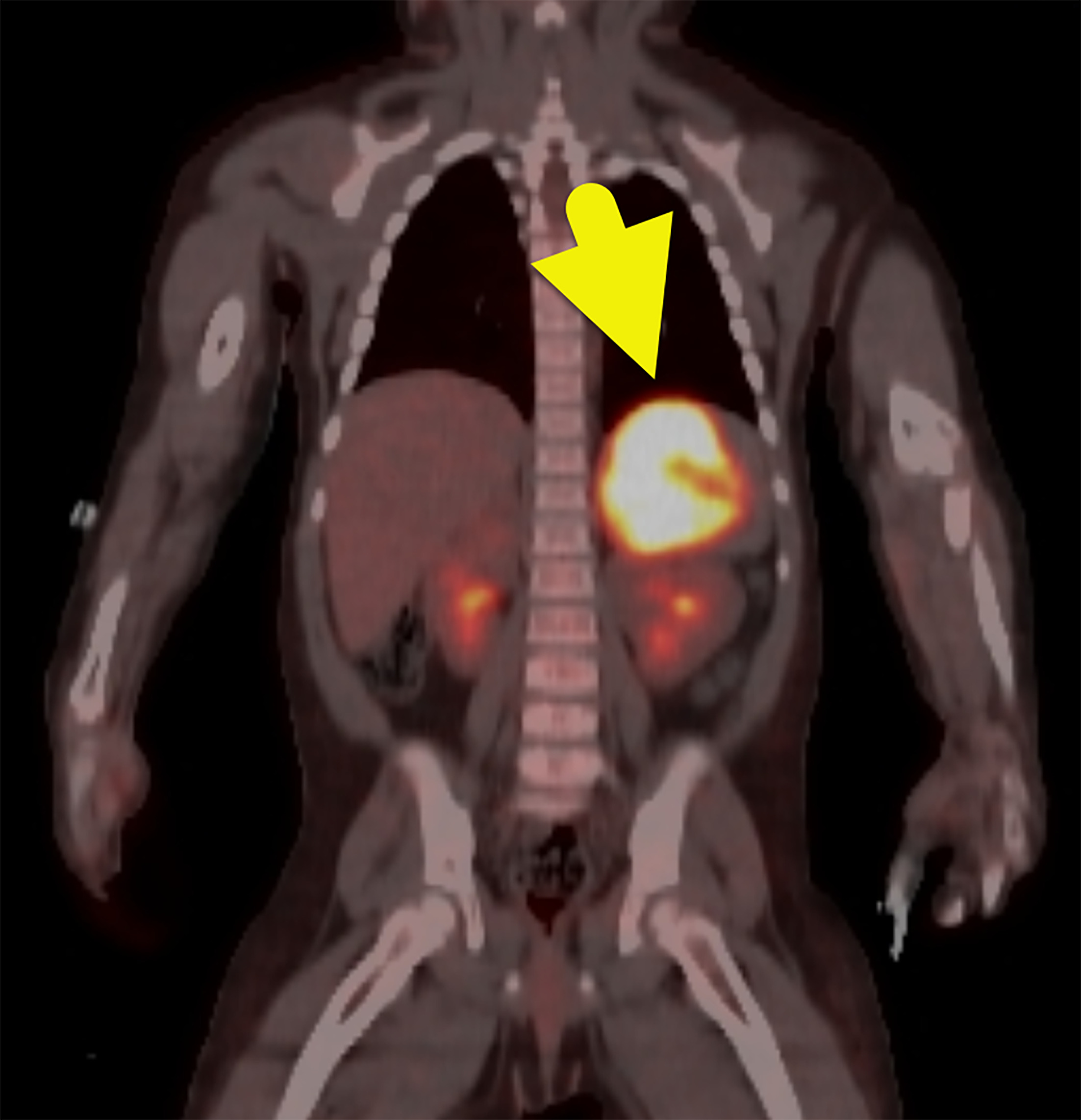
US of the abdomen (Figure 1) revealed a solid left suprarenal mass. An MRI of the abdomen (Figure 2) was performed, which confirmed the mass. It was isointense on T1-weighted images, hyperintense on T2-weighted images, and had minimal enhancement. F-18 fluorodeoxyglucose (FDG) PET-CT (Figure 3) showed the mass to be FDG-avid, with a maximum standardized uptake value of 17.
Diagnosis
Adrenal cortical carcinoma (ACC).
The differential diagnosis of an adrenal mass in young children is ACC, neuroblastoma, ganglioneuroblastoma, ganglioneuroma, Wilms tumor, nephroblastoma, congenital adrenal hyperplasia, and metastases.
Discussion
ACCs are rare, malignant tumors of the adrenal glands, with an incidence estimated to be <1 per 1 million per year. 1 They typically occur within a bimodal age distribution affecting children younger than 5 years of age and adults between 50 and 60 years of age. 2 ACCs are associated with cancer predisposition syndromes in children, including Li-Fraumeni syndrome, Lynch syndrome, Beckwith-Wiedemann syndrome, neurofibromatosis-1, Carney complex, and Werner syndrome. 3 Other syndromes associated with ACC include multiple endocrine neoplasia type 1 and familial adenomatous polyposis. 4
ACCs most commonly occur as a result of sporadic mutations. TP53, a tumor suppressor gene, is the most common somatic mutation in patients diagnosed with ACC. 4 Germline mutations of TP53 at alleles R175H and R337H have led to 10- to 15-fold increases in ACC in communities from southern and southeastern Brazil. 3
ACCs arise from the adrenal cortex. They are typically unilateral tumors, with a median size of 10 cm. 1 Patients may grouped into 1 of 3 different symptomatic classes. The most common symptom class (40-60%) includes patients who present with Cushing syndrome and virilization because of excess adrenal hormone production. 3,5 The second class of patients (about 33%) experience nonspecific symptoms, and the final class of patients (20-30%) are asymptomatic. 3 In children, about 95% of ACCs are functional, while most adult tumors are nonfunctional. 2 Because they are functional, children are typically diagnosed earlier in the course of disease. If found early enough, ACC may remain localized to the adrenal gland. 6 Patients with functional tumors present with increased cortisol, dehydroepiandrosterone-sulfate, urine 17-ketosteroid, and urine 17-hydroxycorticosteroid.
The Weiss score is the most widespread diagnostic tool for assessing ACCs. It is a histologic analysis that uses 9 parameters to evaluate the likelihood of malignancy. 5 A score of 3 or higher is a hallmark of a malignant ACC, although about ~10% of ACCs can score less. 6
Contrast-enhanced CT (CECT), MRI, and FDG-PET/CT are used in the diagnosis and evaluation of ACCs. CECT demonstrates a heterogeneous adrenal mass. While multiphase imaging is often performed in adults to help distinguish adrenal adenoma from ACC, this is not performed in children due to the excess radiation dose associated with multiphase imaging and the rarity of adrenal adenoma in the pediatric population. Contrast- enhanced imaging can highlight central areas of hemorrhage or necrosis.
On MRI, ACCs are hypointense or isointense compared with the liver parenchyma on T1-weighted images and hyperintense on T2-weighted images. 3 After contrast, the tumor has irregular, peripheral, heterogeneous enhancement, with central areas that do not enhance due to hemorrhage or necrosis. 3 On both CT and MRI, the radiologist should look for invasion of the ACC into the inferior vena cava or adjacent organs. 2,3
FDG-PET/CT can identify malignant adrenal tumors and possibly metastases. 2 In adults, FDG-PET/CT can differentiate ACCs from adrenal adenomas with 88-100% specificity and 100% sensitivity. 2,3
There are also 6 characteristic signs from imaging techniques that separate an ACC from benign adenoma or pheochromocytoma. These include a tumor size over 4 cm, tumor shape (irregular, well-demarcated margins), tumor texture (a stellate appearance, internal calcifications, heterogeneous internal enhancement due to hemorrhage, necrosis, and fibrosis), CT x-ray beam attenuation (rarely <10 HU), increased vascularity, and a contrast washout of <50% after 10 minutes of contrast administration. 2,5-7 Image-guided percutaneous biopsy is primarily used to confirm the diagnosis. 8
ACC is an aggressive cancer. The 5-year survival rate in patients below the age of 18 is 46-55%. 9 In adults, the 5-year survival rate declines as the designated stage increases, ranging from 66% to 82% in stage 1 ACC to 0% to 17% in patients with stage 4 disease. 3 The presence of necrosis, a Ki-67 value >10-15% in children (20% in adults), and a Weiss score of at least 6 contribute to a worse prognosis. 3,9,10 With a Weiss score of 6 or higher, the 2-year survival rate in children is decreased to 35%. 9
Treatments for ACC include surgery and chemotherapy. Surgical resection is the mainstay for treating ACC as it reduces glucocorticoids and cortisol levels and debulks or removes the tumor. The benchmark for ACC chemotherapy is platinum therapy (etoposide-doxorubicin-cisplatin) with mitotane. 3 Immunotherapies that block vascular and epidermal growth factors are emerging as new chemotherapy options for ACC as well. 3 Radiation therapy is used for palliation. 3
Conclusion
ACC is an aggressive malignancy that has a poor prognosis because of how late they are discovered. Children with ACC often have an underlying genetic disorder. When a tumor is present, they may present with nonspecific or Cushingoid symptoms. CT, MRI, and FDG-PET are used in diagnosis.
References
- Bancos I, Arlt W. Steroid biomarkers and the diagnosis of adrenal cortical carcinoma. Curr Opin Endocr Metab Res. 2019;8:167-173. doi:10.1016/j.coemr.2019.08.013.
- Gundgurthi A, Kharb S, Dutta M. Childhood adrenocortical carcinoma: case report and review. Indian J Endocr Metab. 2012;16(3):431. doi:10.4103/2230-8210.95699.
- Thampi A, Shah E, Elshimy G, Correa R. Adrenocortical carcinoma: a literature review. Transl Cancer Res. 2020;9(2):1253-1264. doi:10.21037/tcr.2019.12.28.
- Paragliola R, Corsello A, Locantore P. Medical approaches in adrenocortical carcinoma. Biomedicines. 2020;8(12). doi:10.3390/biomedicines8120551.
- Kostiainen I, Hakaste L, Kejo P. Adrenocortical carcinoma: presentation and outcome of a contemporary patient series. Endocrine. 2019;65(1):166-174. doi:10.1007/s12020-019-01918-9.
- Papotti M, Libè R, Duregon E. The weiss score and beyond—histopathology for adrenocortical carcinoma. Horm Cancer. 2011;2(6):333-340. doi:10.1007/s12672-011-0088-0.
- Nakamura Y, Yamazaki Y, Felizola S. Adrenocortical carcinoma: review of the pathologic features, production of adrenal steroids, and molecular pathogenesis. Endocrinol Metab Clin North Am. 2015;44(2):399-410. doi:10.1016/j.ecl.2015.02.007.
- Thabet A, Lahoud R, Shaqdan K, Arellano R, Uppot R. How we do it: adrenal biopsy and ablation. Arab J Intervent Radiol. 2019;3(2):50-57. doi:10.4103/AJIR.AJIR_6_19.
- Sandru F, Petca R, Carsote M. Adrenocortical carcinoma: pediatric aspects (Review). Exp Ther Med. 2022;23(4):287. doi:10.3892/etm.2022.11216.
- Ma C, Xiong J, Su H, Li H. The underlying molecular mechanism and drugs for treatment in adrenal cortical carcinoma. Int J Med Sci. 2021;18(13):3026-3038. doi:10.7150/ijms.60261.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Adrenal Cortical Carcinoma. Applied Radiology. 2025. doi:10.37549/JPCR-25-0051.