Cystic Wilms Tumor
Applied Radiology — Vol. 1 , Issue 1 , pp. 1 -3
Published: September 1, 2025
1 Department of Radiology, University of Cincinnati College of Medicine, Cincinnati, Ohio
2 Department of Radiology, Cincinnati Children’s Hospital, Cincinnati, Ohio
3 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
* Corresponding author: Alexander J. Towbin (alexander.towbin@cchmc.org)
Abstract
Wilms tumor is the most common pediatric renal malignancy. Patients are typically diagnosed at 3-4 years of age. The tumor arises from nephrogenic rests. Approximately 10% of patients with Wilms tumor have an underlying genetic predisposition. Imaging, including CT, MRI, and abdominal ultrasonography, play a crucial role in diagnosing Wilms tumor. Treatment involves a combination of surgery, chemotherapy, and radiation therapy. The prognosis of Wilms tumor is generally good, although relapse can occur in 20% of patients.
Keywords
kidney, neoplasm
Categories
Case Summary
An infant girl with a history of SOFT syndrome (a primordial dwarfism condition characterized by short stature, onychodysplasia, facial dysmorphism, and hypotrichosis) presented to another hospital with bilateral renal masses.
Imaging Findings
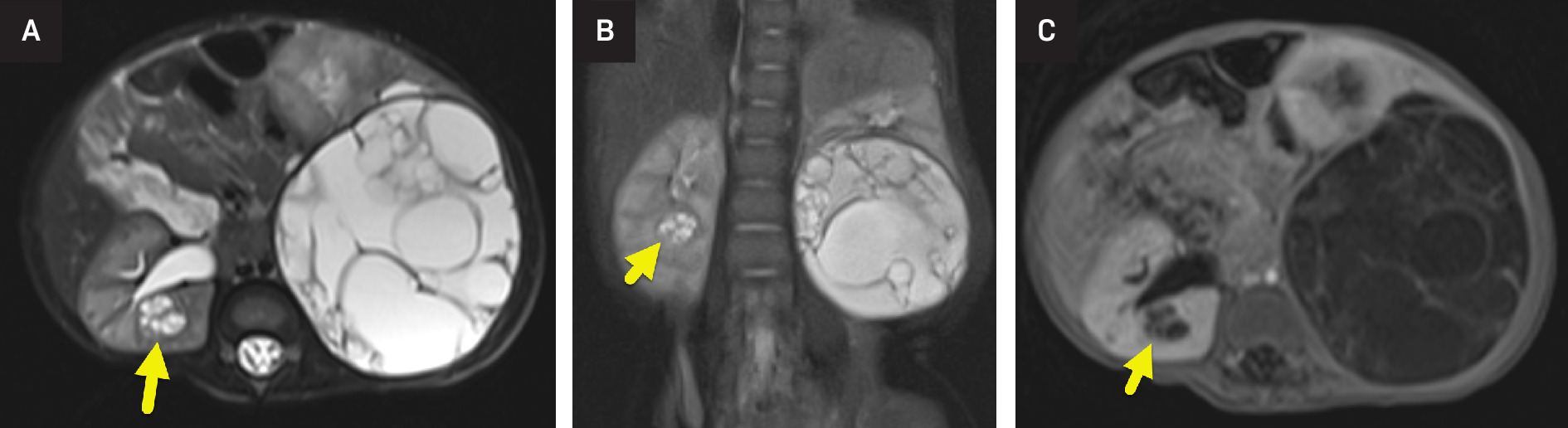
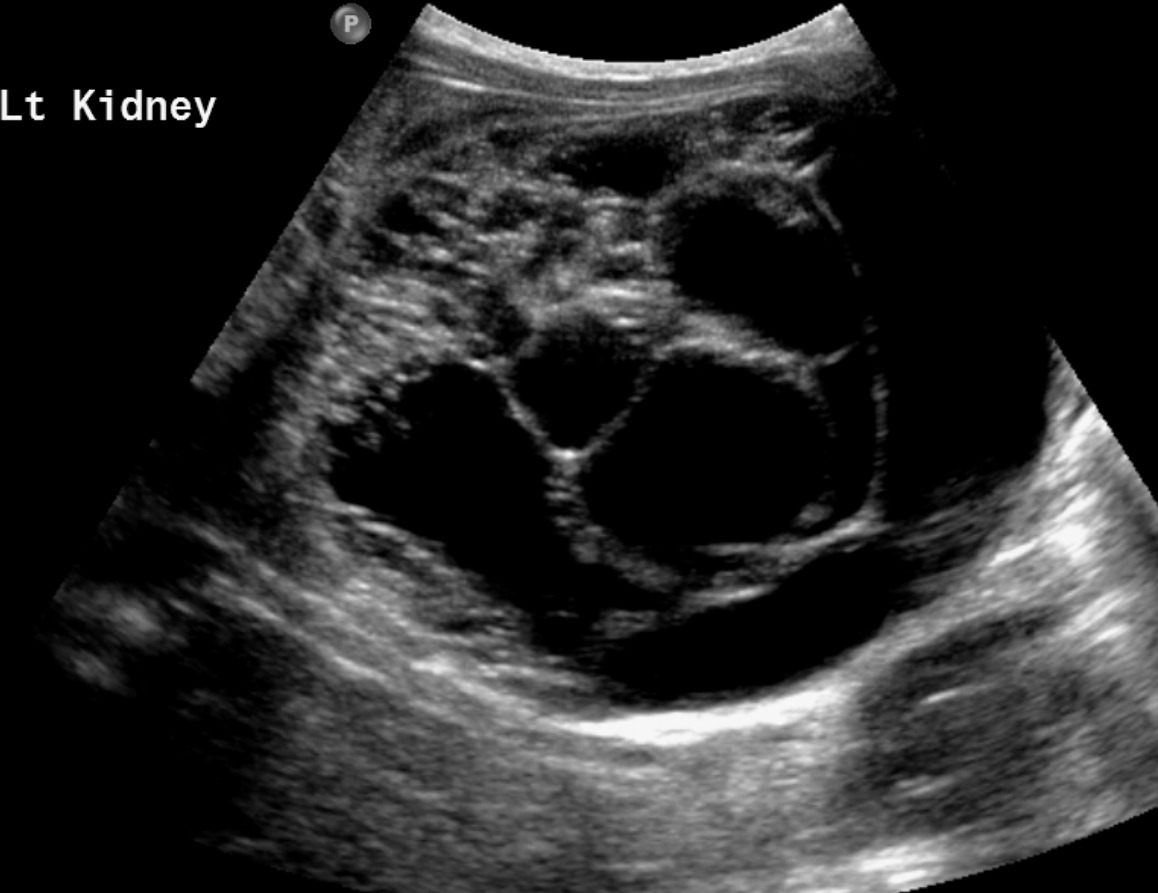
Abdominal MRI (Figure 1) showed multiple large, multiseptate cystic masses involving the left kidney. The largest mass measured 7 × 6.2 cm and followed fluid signal on all sequences. There was minimal enhancement of the fine septations. Several smaller masses were present within the right kidney. Masses in both kidneys abutted but did not extend into the renal pelvis. An US (Figure 2) highlighted the multiseptated cystic renal mass.
Diagnosis
Cystic Wilms tumor.
The imaging differential diagnosis includes multilocular cystic nephroma.
Discussion
Wilms tumor is the most common renal malignancy of childhood and the most common solid pediatric abdominal tumor. It occurs in young children, diagnosed at a mean age of 3-4 years. Affected patients may present with a palpable abdominal mass, abdominal pain, hematuria (gross or microscopic), fever, and hypertension. Wilms tumor arises from persistent embryologic structures. Embryologically, the fetal kidney develops from the ureteric bud and the metanephric blastema. Typically, the metanephric blastema involutes by the 36th week of gestation. Any residual metanephric blastema is termed a nephrogenic rest. 1 It is estimated that nephrogenic rests play a role in the development of 28-40% of solitary and 90-100% of bilateral Wilms tumors. 2
Most Wilms tumors occur randomly and are solitary at presentation. However, 10% of patients have synchronous unilateral tumors, and 5-7% have synchronous bilateral tumors. Patients with an underlying Wilms tumor predisposition syndrome are more likely to have multifocal or bilateral tumors. 3
Approximately 15% of patients with Wilms tumor have an underlying genetic mutation that increases the risk of cancer. The WT1 gene, located on chromosome 11p13, was one of the first genes identified in the development of the tumor. Other genes where mutations are associated with Wilms tumor include CTNNB, AMER1, IGF2, and H19. In addition to somatic mutations, Wilms tumor is associated with predisposition syndromes. These syndromes are classified into 3 categories based on the risk of developing the tumor: high (>20%), moderate (5-20%), and low (<5%). Some of the high-risk syndromes include WAGR (Wilms tumor, aniridia, genitourinary anomalies, and intellectual disability), Denys-Drash syndrome, Perlman syndrome, and Fanconi anemia. Moderate-risk syndromes include Frasier syndrome and Beckwith-Wiedemann syndrome. Finally, low-risk syndromes include Bloom syndrome, Li-Fraumeni syndrome, and Mulibrey nanism. 4 SOFT syndrome, also known as POC1A-related (vPOC1A) syndrome, is a rare genetic condition that is inherited in an autosomal-recessive manner. It is not known to be associated with Wilms tumor. 5
Screening is offered to children with a cancer predisposition syndrome, such as Beckwith-Wiedemann syndrome. Renal US is the preferred screening technique due to its low cost, high spatial resolution, lack of radiation, and ability to be performed in infants without the need for sedation. US is recommended to be performed every 3 months until the child is 7 years of age when the risk of developing Wilms tumor is significantly reduced. The objective of Wilms tumor screening is to detect the condition early so that less toxic chemotherapy can be used, renal-sparing surgery can be performed, and radiation therapy can be avoided. Patients with predisposition syndromes may develop a metachronous tumor in the other kidney. Thus, this treatment strategy helps maintain maximal kidney function while ensuring oncological control. 6
Imaging plays a crucial role in the diagnostic work-up of children with a renal mass. Abdominal radiograph may be the first imaging study performed. The radiograph may show a soft-tissue mass that displaces loops of the bowel away from the tumor. Abdominal ultrasonography is typically performed next and used to confirm the presence of the mass and determine its organ of origin. When interpreting an US, the radiologist should look to identify the tumor’s effect on the renal parenchyma, the renal collecting system, the renal vein, and the inferior vena cava. Once the tumor is confirmed to arise from the kidney, cross-sectional imaging should be performed. The most common sites of metastatic disease in Wilms tumor are the lungs, liver, extra-abdominal nodes, brain, and bone. 7
CT and MRI (Figure 1) are similar in their ability to diagnose and stage Wilms tumor. Benefits of CT include its speed, availability, and ability to image the chest and abdomen in a single setting. MRI offers excellent soft-tissue resolution and avoids abdominal radiation. It may also be more sensitive for the detection of small nephrogenic rests. 6 On imaging, Wilms tumor most commonly appears as a solid, heterogeneous mass visibly separated from surrounding renal tissue by a fibrous capsule. On both CT and MR, the mass enhances less than the adjacent kidney. Calcification is present in approximately 15% of tumors and is seen more easily on CT or US (Figure 2). 8 While cystic tumors are possible, they have only rarely been reported. During the assessment of Wilms tumors, the radiologist should assess the renal vein and inferior vena cava as both may be invaded. 8,9
Multilocular cystic nephroma is the most common cystic renal mass in young children. It is typically impossible to distinguish a multilocular cystic nephroma from a cystic Wilms tumor by imaging. Thus, all cystic masses are resected. Features that support a diagnosis of multilocular cystic nephroma include herniation of the mass into the renal pelvis and a known diagnosis of DICER1 gene mutation. Features that support a diagnosis of a cystic Wilms tumor include irregular and/or nodular septations within the mass and nephrogenic rests. 1,10
Wilms tumor is staged based on the extent of the tumor at the time of diagnosis and resection. It is divided into 5 stages:
-
Stage 1: Tumor is limited to the kidney and is completely resected by surgery. There may be involvement of intrarenal vessels but no involvement of renal sinus vessels.
-
Stage 2: Tumor has grown beyond the kidney, either into the renal sinus or blood and lymphatic vessels, but it was removed completely by surgery without any cancer left behind.
-
Stage 3: Tumor is assigned to stage 3 for any of the following reasons: (1) the tumor extends beyond the resection margins or there is visible residual disease; (2) cancer is present in abdominal lymph nodes; (3) the tumor ruptures before or during surgery; (4) the tumor is resected in piecemeal; and (5) any biopsy performed prior to surgery.
-
Stage 4: Distant metastases are present.
-
Stage 5: Tumors are found in both kidneys at the time of initial diagnosis.1
In general, Wilms tumor is treated with surgery, chemotherapy, and radiation therapy. The exact treatment plan depends on the stage of the disease and other factors such as the patient’s age, overall health, and the size and location of the tumor.
In stage 1 and 2 Wilms tumors, upfront surgery is performed, followed by chemotherapy. In stage 3 and 4 tumors, neoadjuvant chemotherapy is used prior to resection. Radiation therapy is also provided during therapy. 11 In patients with bilateral disease, preoperative chemotherapy is followed by renal-sparing surgery to try to cure the disease while maintaining the highest level of kidney function possible. 4
The overall prognosis of Wilms tumor is good but varies based on tumor histology and stage, response to preoperative chemotherapy, and presence of lung metastases. Patients with stage 4 anaplastic Wilms tumor or blastemal-type Wilms tumor have a relatively poor prognosis, with an overall survival rate of <50%.
Surveillance with abdominal US and chest radiograph is recommended after the completion of therapy. Approximately 20% of patients with Wilms tumor will relapse. When patients relapse, the overall survival is approximately 50%. 6
Conclusion
Wilms tumor is the most common pediatric renal malignancy. Patients are typically diagnosed at 3-4 years of age. The tumor arises from nephrogenic rests. Approximately 10% of patients with Wilms tumor have an underlying genetic predisposition. Imaging, including CT, MRI, and abdominal ultrasonography, plays a crucial role in diagnosing Wilms tumor. Treatment involves a combination of surgery, chemotherapy, and radiation therapy. The prognosis of Wilms tumor is generally good, although relapse can occur in 20% of patients.
References
- Szychot E, Apps J, Pritchard-Jones K. Wilms’ tumor: biology, diagnosis and treatment. Transl Pediatr. 2014;3(1):12-24. doi:10.3978/j.issn.2224-4336.2014.01.09.
- Sandberg J, Chi Y, Smith E. Imaging characteristics of nephrogenic rests versus small wilms tumors: a report from the children’s oncology group study AREN03B2. AJR Am J Roentgenol. 2020;214(5):987-994. doi:10.2214/AJR.19.22301.
- Artunduaga M, Eklund M, van der Beek J. Imaging of pediatric renal tumors: a COG diagnostic imaging committee/SPR oncology committee white paper focused on Wilms tumor and nephrogenic rests. Pediatr Blood Cancer. 2023;70 Suppl 4(suppl 4). doi:10.1002/pbc.30004.
- Treger T, Chowdhury T, Pritchard-Jones K, Behjati S. The genetic changes of Wilms tumour. Nat Rev Nephrol. 2019;15(4):240-251. doi:10.1038/s41581-019-0112-0.
- Li G, Chang G, Wang C. Identification of SOFT syndrome caused by a pathogenic homozygous splicing variant of POC1A: a case report. BMC Med Genomics. 2021;14(1):1-5. doi:10.1186/S12920-021-01055-1/FIGURES/3.
- Spreafico F, Fernandez C, Brok J. Wilms tumour. Nat Rev Dis Primers. 2021;7(1):75. doi:10.1038/s41572-021-00308-8.
- Pater L, Melchior P, Rübe C. Wilms tumor. Pediatr Blood Cancer. 2021;68 Suppl 2(s2). doi:10.1002/pbc.28257.
- Kurose N, Takenaka M, Yamashita M. A case report of infantile cystic nephroblastoma. Diagn Pathol. 2018;13(1):1-5. doi:10.1186/S13000-018-0761-5/FIGURES/4.
- Kaste S, Dome J, Babyn P. Wilms tumour: prognostic factors, staging, therapy and late effects. Pediatr Radiol. 2008;38(1):2-17. doi:10.1007/s00247-007-0687-7.
- Xu L, Wang L, Tian X, Gai Y, Shang N. Spontaneously ruptured multilocular cystic nephroma in an infant. J Clin Ultrasound. 2023;51(1):107-109. doi:10.1002/jcu.23267.
- Wilms tumor and other childhood kidney tumors treatment (PDQ®)–health professional version - NCI.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Cystic Wilms Tumor. Applied Radiology. 2025;1(1):1-3. doi:10.37549/JPCR-25-0004.