Hypertrophic Cardiomyopathy
Applied Radiology
Published: January 27, 2026
1 Kansas City University College of Medicine, Kansas City, Missouri
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 Heart Institute, LeBonheur Children’s Hospital, Memphis, Tennessee
4 Department of Radiology, Cincinnati Children’s Hospital, University of Cincinnati College of Medicine, Cincinnati, Ohio
Abstract
Hypertrophic cardiomyopathy (HCM) is an inheritable cardiac disease characterized by the hypertrophy of the myocardial segments.HCM is the leading cause of sudden cardiac death in young athletic adults in the United States. While the prevalence rate of HCM is 0.2-0.8%, it is widely underdiagnosed. Left ventricle hypertrophy is most observed, but other myocardial segments are also susceptible to hypertrophy. These include anterolateral wall, apex, mid-ventricular, isolated basal septa, and asymmetric interventricular septal hypertrophy. Outcomes in HCM have improved over the years but continue to require access to better therapies in young children, who currently do not have access to myosin inhibitors.
Keywords
cardiac, genetic, myocardial
Categories
Case Summary
A preschool-aged boy with a family history of sudden death of unknown etiology presented with chest pain and palpitations. His physical exam was notable for normal vital signs, but a II/VI systolic ejection murmur was present at the left sternal border that increased in intensity on Valsalva maneuver.
Imaging Findings
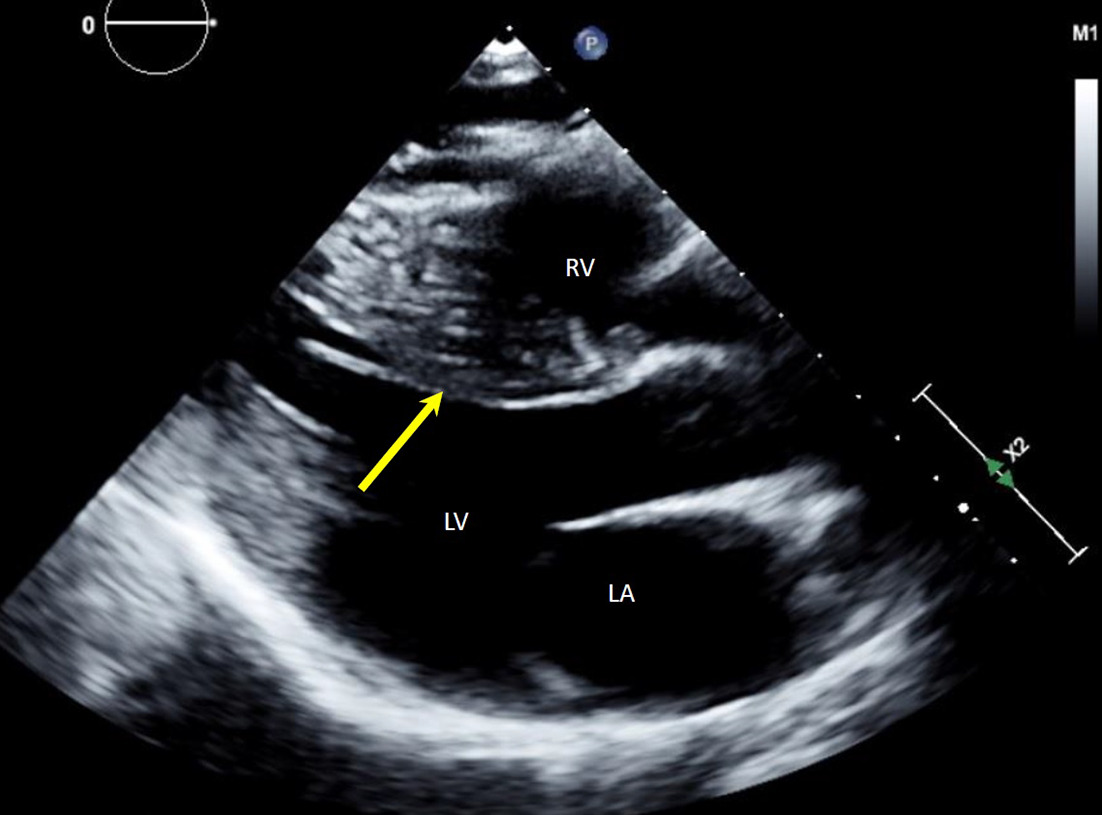
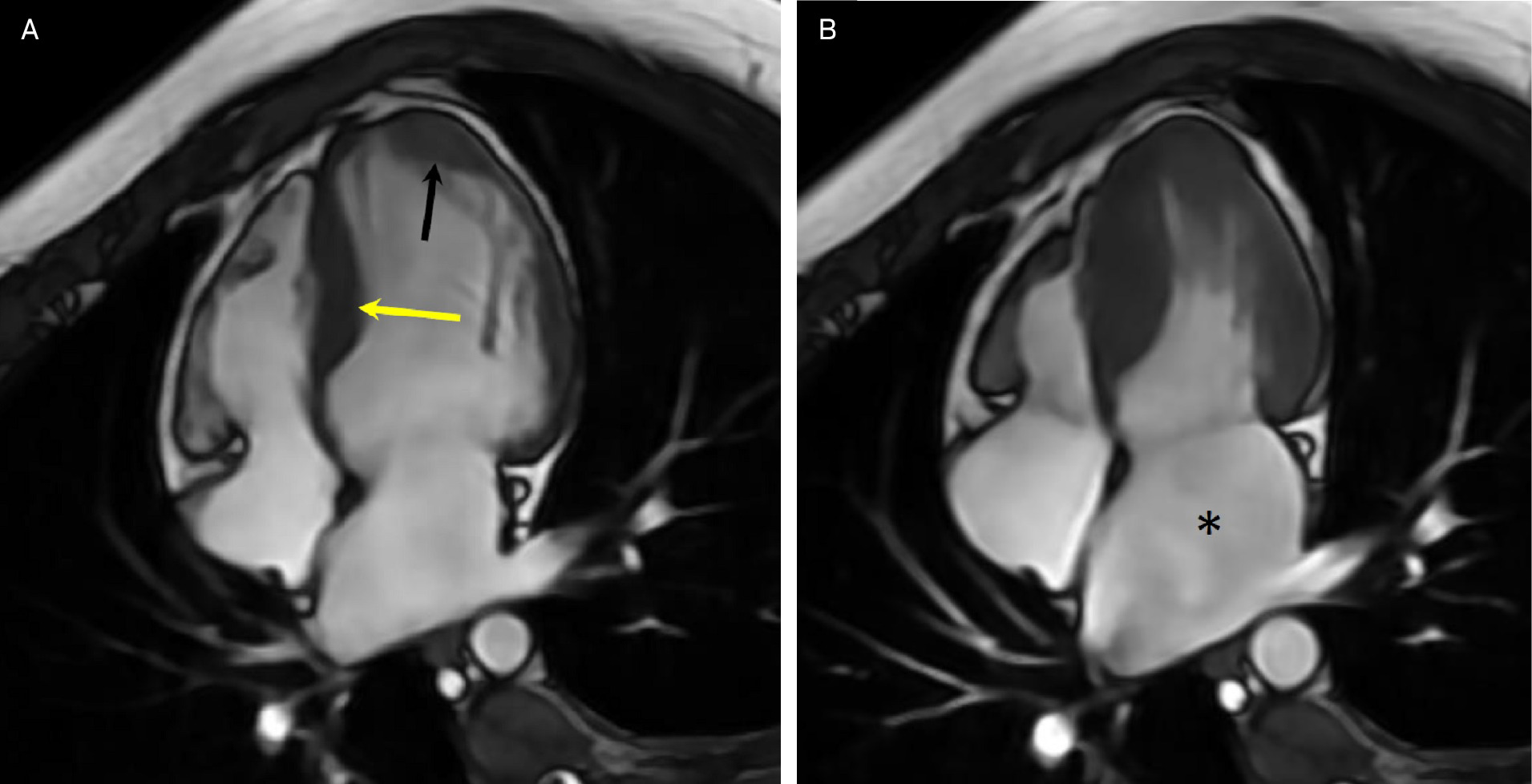
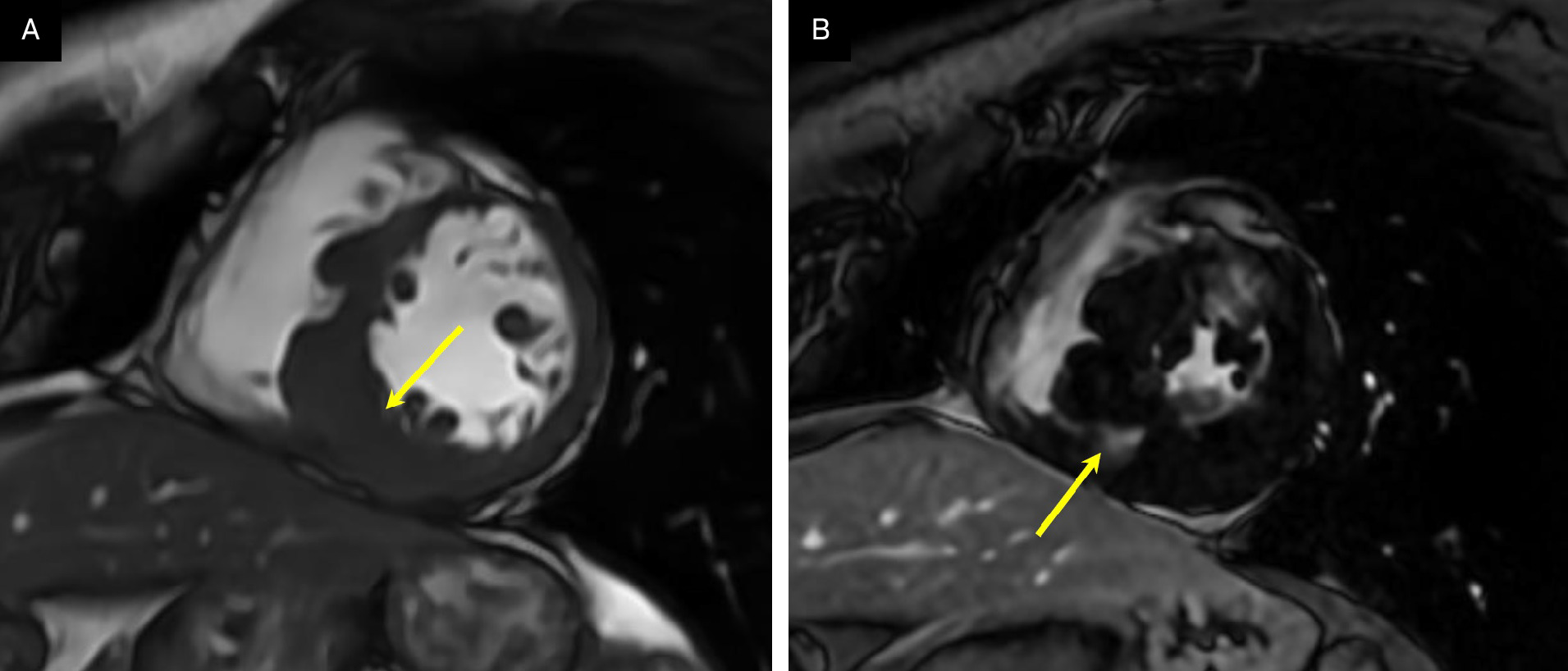
Transthoracic echocardiogram (Figure 1) showed severe asymmetric left ventricular hypertrophy (LVH) with normal systolic function. Cardiovascular magnetic resonance (CMR) confirmed severe asymmetric LVH, with the mid-septal wall measuring 2.5 cm in diameter (normal 1-1.5 cm) with normal systolic function and severely dilated left atrium consistent with restrictive physiology (Figure 2). There was extensive myocardial fibrosis present (Figure 3) with a patchy mid-myocardial late gadolinium enhancement (LGE) pattern of the mid-septal wall.
Diagnosis
Hypertrophic cardiomyopathy (HCM).
The differential diagnosis of HCM includes causes such as idiopathic HCM, aortic stenosis, coarctation of the aorta, coarctation, chronic systemic hypertension, or athletic heart syndrome. Alternatively, genetic or associated diseases such as amyloidosis, Fabry disease, Pompe disease, dilated cardiomyopathy, and restrictive cardiomyopathy.
Discussion
HCM is an inheritable cardiac disease characterized by the hypertrophy of the myocardial segments. 1 HCM is the leading cause of sudden cardiac death (SCD) in young athletic adults in the United States. While the prevalence rate of HCM is 0.2-0.8%, it is widely underdiagnosed. In children <1 year old, HCM may be associated with diastolic and/or systolic dysfunction and most commonly is caused by metabolic-based abnormalities or syndromes (mitochondrial dysfunction, Pompe disease, Noonan syndrome, etc). However, in young adults, the disorder is mostly associated with primary diastolic dysfunction. 1
Left ventricular outflow tract or mid-ventricular obstruction may occur. In cases with severe diastolic dysfunction, a restrictive physiology with a dilated left atrium and no mitral regurgitation may develop, and these patients are the highest risk individuals. Nonsarcomeric causes such as RASopathies like Noonan syndrome, metabolic storage disorders, neurological illnesses, and mitochondrial disorders account for the majority of the HCM cases in infants under 1 year of age and neonates. 2 HCM due to mutations in sarcomeric genes is most common in ages 1-18 and adults. The most common sarcomeric gene mutations involved in HCM are the β-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) gene, accounting for nearly 70% of all HCM cases. 2 Neonates of mothers with diabetes are also at high risk for developing HCM due to reactive hyperinsulinemia, which typically resolves within the first weeks of life. 3 Additionally, neonatal patients with severe congenital hyperinsulinemia are also at risk. In such cases, a pancreatectomy is needed to treat hyperinsulinemia and resolve the HCM. 3 Histopathological findings in pediatric and young adult patients with HCM include myocyte disarray, abnormal branching, and woven interstitial fibrosis. The myocardial cell diameter commonly ranges from 20 to 30 μm, indicating mild to severe HCM. 4
LVH is most observed, but other myocardial segments are also susceptible to hypertrophy. These include anterolateral wall, apex, mid-ventricular, isolated basal septa, and asymmetric interventricular septal hypertrophy. 5 In the neonatal population, histopathological findings include cardiomyocyte nuclei enlargement with mild fibrosis and diffuse concentric ventricular hypertrophy. 6 Children and young adults with HCM may present with angina, palpitations, syncope with exercise, lightheadedness, dyspnea, or SCD. In patients with suspected HCM, a comprehensive cardiac history, 3-generation family history, echocardiogram, CMR, stress echocardiography, 12-lead ECG, and genetic testing are recommended. 7 Key echocardiographic features suggestive of HCM include increased left ventricular wall thickness of >15 mm in adults or a Z-score >2.0 in children, mitral valve leaflet elongation, and anterior displacement of the anterolateral and posteromedial papillary muscles of the mitral valve. 8 A dynamic left ventricular outflow tract obstruction (LVOTO) may be observed due to the systolic anterior motion of the mitral valve leaflets in patients with obstructive HCM. 8 Advances in genomic medicine have strengthened the echocardiographic-genotype relationship. Zhou et al demonstrated that mutations in MYH7 and MYBPC3 correlate with greater susceptibility to myocardial thickening of the anterior wall, interventricular septum, and lateral wall. Mutations in the ALPK3 correlate with apex hypertrophy. 9 Diagnosing and managing HCM often requires CMR for optimal imaging, particularly in distinguishing hypertrophy of different myocardial segments. CMR also enhances the characterization of the mitral valve and papillary muscle anomalies, which are crucial for managing obstructive HCM. 5 CMR’s ability to further accurately delineate the initial echocardiography findings may be essential in evaluating LVOTO mechanisms and septal reduction therapy planning. 5
Moreover, LGE in CMR can monitor HCM by assessing the extent of cardiac fibrosis. 5 A CMR of adult and pediatric patients may reveal extensive LGE, which strongly correlates with a higher incidence of SCD. 5 Given its comprehensive evaluation capabilities, CMR should be integrated into the diagnostic and therapeutic decisions involving implantable cardioverter defibrillator placement. Outcomes in HCM have improved over the years but continue to require access to better therapies in young children, who currently do not have access to myosin inhibitors. Despite having tragic consequences, HCM carries a favorable prognosis if diagnosed early. Managing the symptoms includes negative inotropic drugs such as β-blockers, myosin inhibitors such as mavacamten and afikomen (pending FDA approval), anti-arrhythmics, and anticoagulants. Prophylactic management may entail placing an implantable cardioverter defibrillator for SCD prevention. Long-term management may involve surgical myectomy, rarely alcohol septal ablation, and heart transplantation in end-stage progressive heart failure due to hypertrophic obstructive cardiomyopathy, or nonobstructive HCM with systolic dysfunction, or HCM with restrictive physiology. 10
References
- Towbin J. Hypertrophic cardiomyopathy. Pacing Clin Electrophysiol. 2009;32(s2):S23-S31. doi:10.1111/j.1540-8159.2009.02381.x.
- Lee T, Hsu D, Kantor P. Pediatric cardiomyopathies. Circ Res. 2017;121(7):855-873. doi:10.1161/CIRCRESAHA.116.309386.
- Huang T, Kelly A, Becker S, Cohen M, Stanley C. Hypertrophic cardiomyopathy in neonates with congenital hyperinsulinism. Arch Dis Child Fetal Neonatal Ed. 2013;98(4):F351-F354. doi:10.1136/archdischild-2012-302546.
- Tejado B, Jou C. Histopathology in HCM. Glob Cardiol Sci Pract. 2018;2018(3):20. doi:10.21542/gcsp.2018.20.
- Brenes J, Doltra A, Prat S. Cardiac magnetic resonance imaging in the evaluation of patients with hypertrophic cardiomyopathy. Glob Cardiol Sci Pract. 2018;2018(3):22. doi:10.21542/gcsp.2018.22.
- Paauw N, Stegeman R, De Vroede M. Neonatal cardiac hypertrophy: the role of hyperinsulinism-a review of literature. Eur J Pediatr. 2020;179(1):39-50.
- Maron B, Desai M, Nishimura R. Diagnosis and evaluation of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2022;79(4):372-389. doi:10.1016/j.jacc.2021.12.002.
- Mandeş L, Roşca M, Ciupercă D, Popescu B. The role of echocardiography for diagnosis and prognostic stratification in hypertrophic cardiomyopathy. J Echocardiogr. 2020;18(3):137-148. doi:10.1007/s12574-020-00467-9.
- Zhou N, Weng H, Zhao W. Gene-echocardiography: refining genotype-phenotype correlations in hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2023;25(1):127-135. doi:10.1093/ehjci/jead200.
- Maron B, Desai M, Nishimura R. Management of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2022;79(4):390-414. doi:10.1016/j.jacc.2021.11.021.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Hypertrophic Cardiomyopathy. Applied Radiology. 2026. doi:10.37549/JPCR-25-0061.