Juvenile Dermatomyositis
Journal of Pediatric Case Reports — Vol. 1 , Issue 3
Published: April 1, 2026
1 Texas College of Osteopathic Medicine, Fort Worth, Texas
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 Department of Radiology, Cincinnati Children’s Hospital, University of Cincinnati College of Medicine, Ohio
* Corresponding author: Richard B. Towbin (rtowbin@gmail.com)
Abstract
Juvenile dermatomyositis is a rare autoimmune inflammatory myopathy characterized by progressive proximal muscle weakness and distinctive skin manifestations. Imaging, particularly MRI, plays a crucial role in diagnosis and monitoring, detecting muscle edema, inflammation, and chronic atrophy. Calcinosis is a common complication, particularly in children, and serves as a marker of disease progression. Treatment primarily involves glucocorticoids, with immunosuppressive agents used to minimize long-term steroid exposure. While remission is achievable, ongoing management is necessary to prevent relapses and complications.
Keywords
musculoskeletal, myositis, skin, chronic, cause unknown
Categories
Case Summary
A child with long-standing undertreated dermatomyositis was imaged to assess soft tissue complications.
Imaging Findings
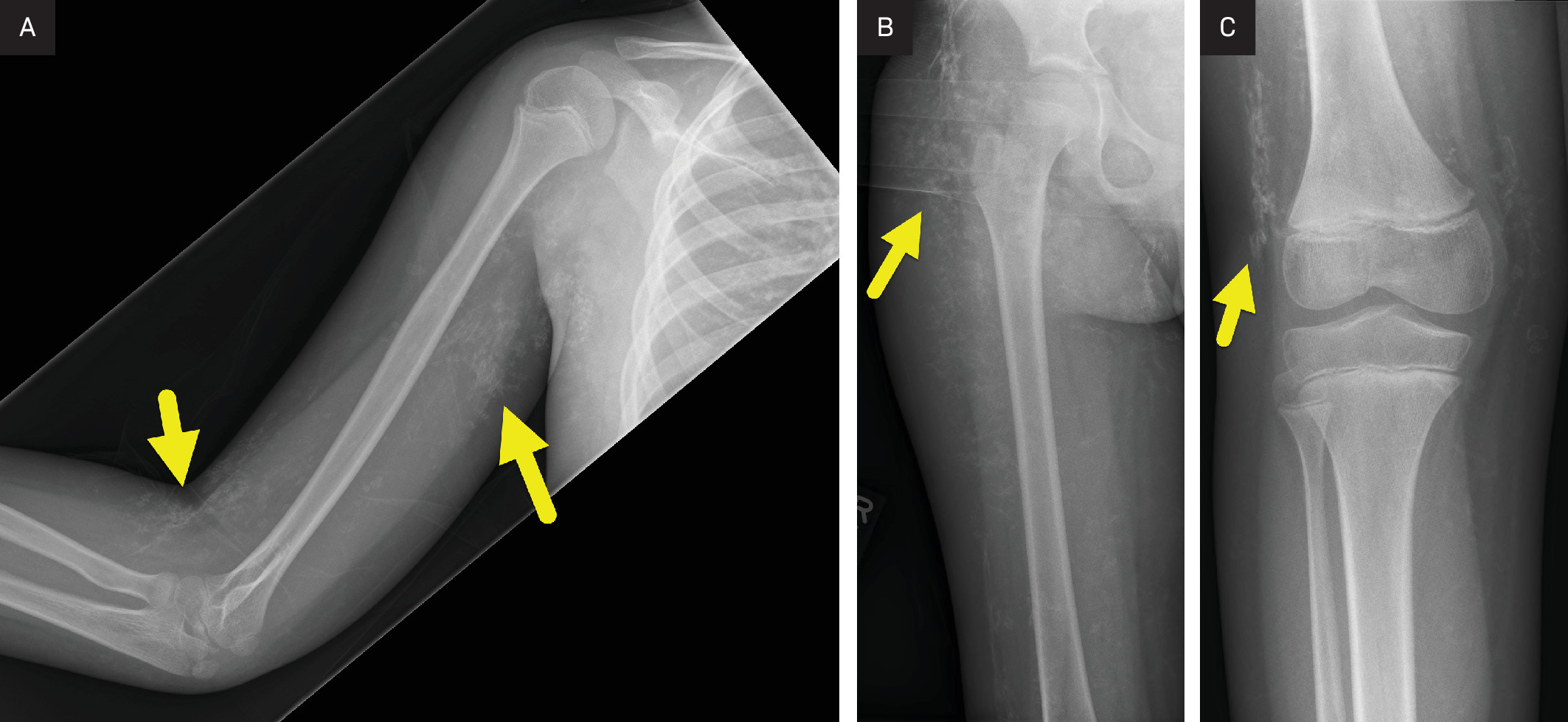
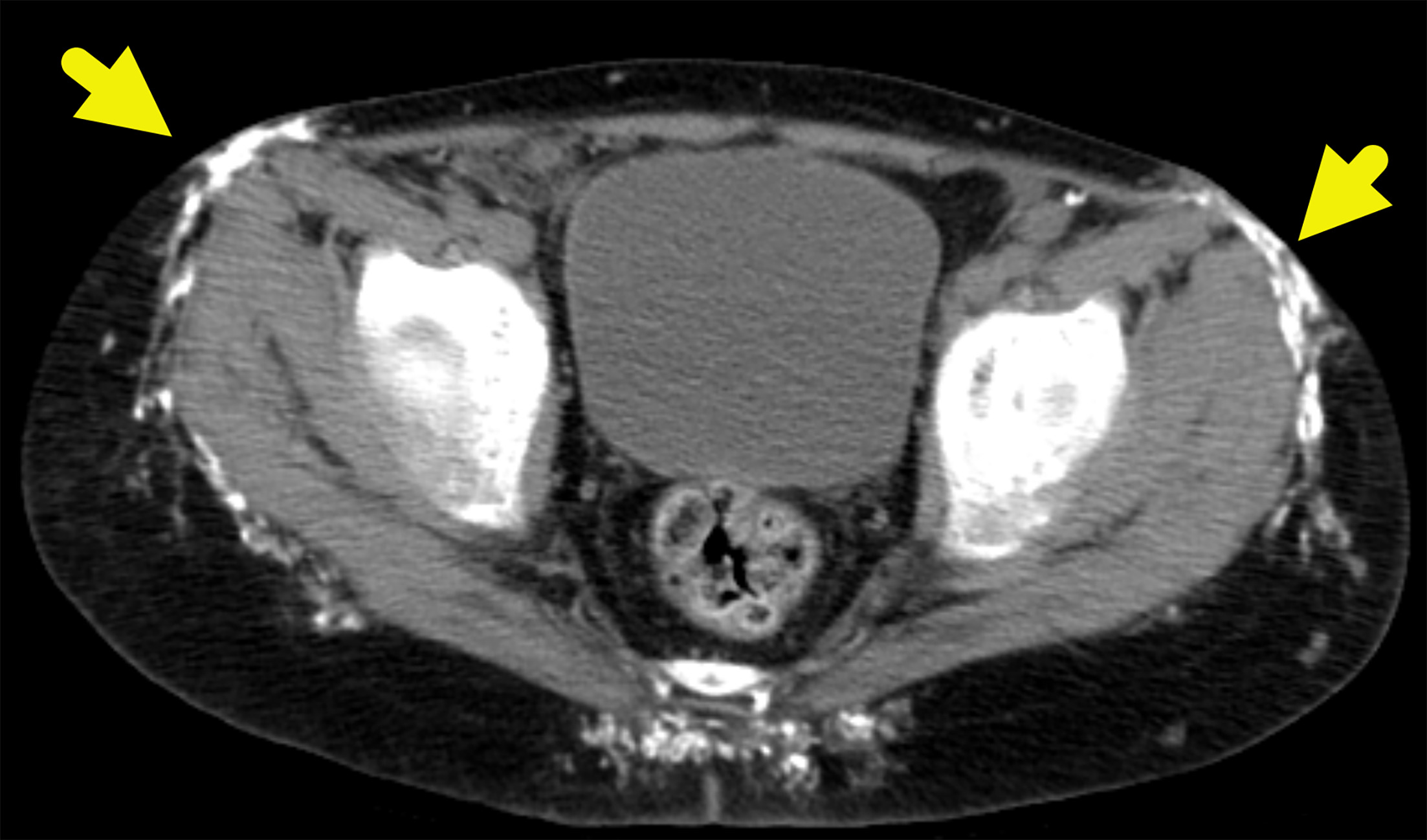
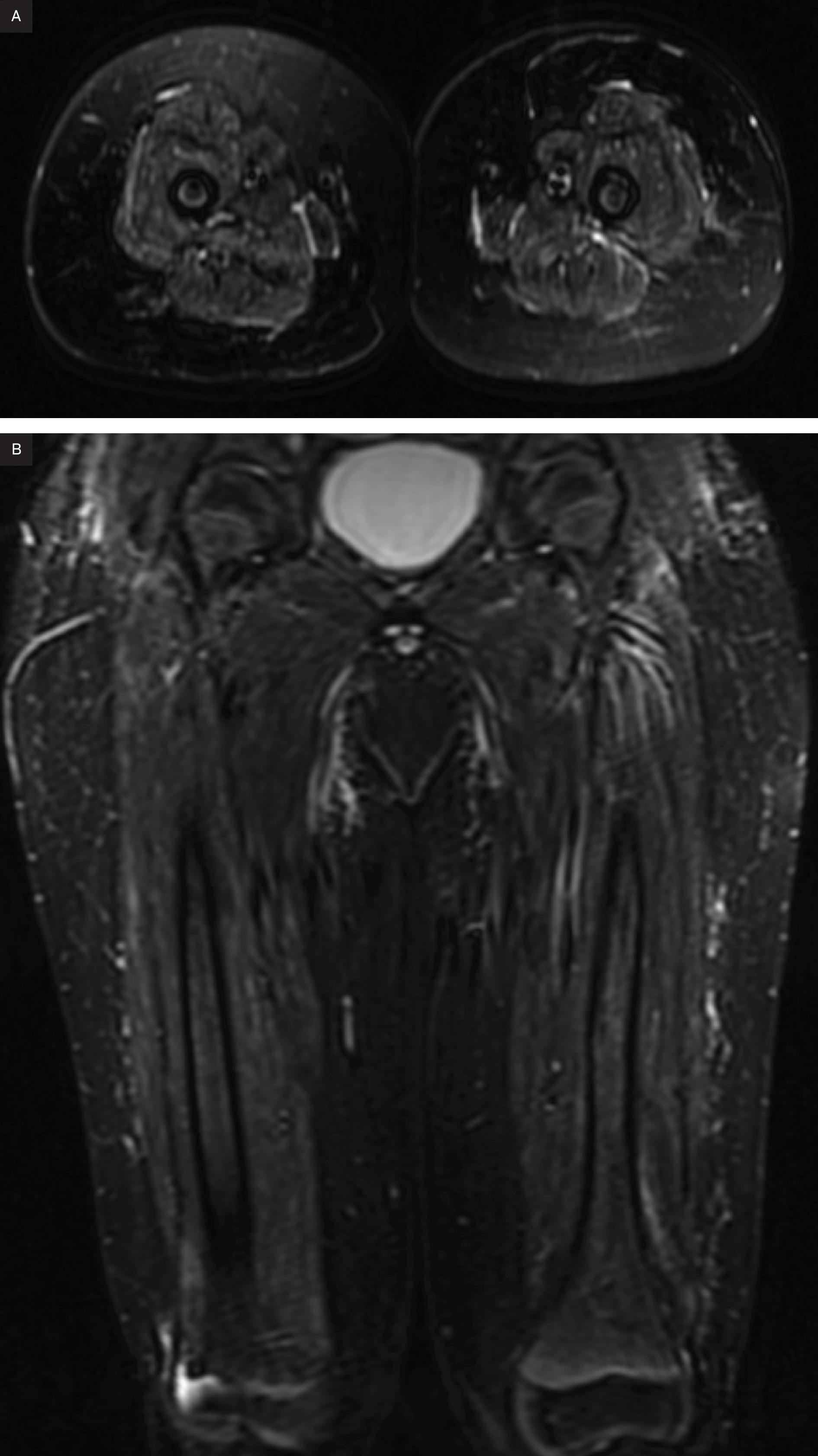
Radiographs (Figure 1) of the upper and lower extremity and CT of the pelvis (Figure 2) show extensive subcutaneous soft tissue calcifications (arrows). MRI at the level of the acetabulum and lower extremity showing calcification and muscle involvement. (Figure 3)
Diagnosis
Dermatomyositis.
The clinical differential diagnosis includes various types of myopathies, including mitochondrial myopathies, infectious and inflammatory myopathies, autoimmune necrotic myopathies plus Duchenne or Becker muscular dystrophy, systemic lupus erythematosus, and juvenile idiopathic arthritis. Polymyositis, systemic sclerosis, and mixed connective tissue disease may have similar imaging profiles.
Discussion
Dermatomyositis is a rare autoimmune inflammatory muscle disorder of unknown cause that primarily affects skeletal muscle and skin. The incidence is estimated at 9.63 per million adults and 1.9‐4.1 per million children.1,2 When dermatomyositis occurs in children, it is referred to as juvenile dermatomyositis. The juvenile form has distinct clinical and laboratory findings compared with the adult form. Characteristic features include progressive muscle weakness and skin rashes, often accompanied by systemic symptoms such as anorexia, pain, fever, irritability, and general deterioration.3 Compared with adult-onset disease, juvenile dermatomyositis is associated with a lower malignancy risk and a more consistent response to therapy.
Juvenile dermatomyositis is classified as an inflammatory myopathy, a group of uncommon conditions characterized by chronic muscle inflammation and weakness.1 While myopathy is a broad term encompassing various muscle disorders, juvenile dermatomyositis can be distinguished through a combination of clinical symptoms, laboratory findings, histopathologic evaluation, and imaging.
Juvenile dermatomyositis typically presents in children 5-14 years of age with a subacute onset of muscle weakness, primarily affecting proximal muscles symmetrically, like adult dermatomyositis. Routine activities such as brushing teeth, climbing stairs, lifting objects, and dressing become progressively difficult.
While distal muscle weakness, pain, and stiffness are rare in adults, they occur more frequently in children, making it more challenging to distinguish juvenile dermatomyositis from juvenile idiopathic arthritis.4 Dysphagia or dysphonia is reported in 18-20% of patients.5 On examination, proximal muscle weakness is the most prominent finding, while muscle tenderness is typically mild, and distal strength remains preserved unless the disease is severe and longstanding.
When a muscle biopsy is performed, findings diagnostic of dermatomyositis include perivascular and perimysial inflammatory infiltrates composed of B cells, CD4+ T helper cells, macrophages, and plasmacytoid dendritic cells, along with perifascicular atrophy and microangiopathy.1
In both children and adults with dermatomyositis, characteristic skin lesions often precede muscle weakness. In up to 40% of patients, skin involvement is the sole presenting feature, a condition known as dermatomyositis sine myositis.
Dermatomyositis is associated with distinctive cutaneous manifestations, including the heliotrope rash—a violaceous discoloration of the eyelids—and Gottron’s papules, which are pathognomonic for the disease.6 Gottron’s papules present as violaceous papules over the metacarpophalangeal joints, proximal and distal interphalangeal joints of the hands, and sometimes on the extensor surfaces of the elbows and knees. Additional findings include periungual erythema with telangiectasia, cuticular hypertrophy, facial edema, and a V-shaped erythematous patch over the anterior neck and upper chest. Raynaud phenomenon may also be present.3
Histopathologic findings in dermatomyositis closely resemble those seen in systemic lupus erythematosus. Key features include vacuolar changes in the basal layer, increased lymphocytic infiltration, and elevated mucin deposition within the dermis. Due to overlapping clinical and histologic features with other autoimmune diseases, a comprehensive evaluation is necessary before establishing a definitive diagnosis.1
Although the diagnosis of dermatomyositis is primarily based on clinical, biochemical, and histopathological findings, imaging plays an important role in disease evaluation. It aids in identifying calcinosis, detecting muscle changes in the active disease phase, diagnosing complications, ruling out other conditions, guiding biopsies, and monitoring disease progression during follow-up.7
Calcinosis, or dystrophic soft tissue calcifications, develops in 30-70% of children with juvenile dermatomyositis and typically occurs in the chronic phase of the disease. These calcium deposits are more common in children and adolescents and form in areas of skin, subcutaneous tissue, fascial planes, and muscle necrosis. In juvenile dermatomyositis, calcinosis can develop within 6 months of disease onset and progressively worsens over time. Because the extent of calcinosis correlates with disease duration, it serves as an important marker of disease progression, alongside muscle weakness and elevated creatine kinase and lactate dehydrogenase levels.8 MRI is the preferred imaging modality for diagnosing and monitoring dermatomyositis, offering detailed visualization of both superficial and deep tissues. Unlike US, MRI is not operator-dependent, provides a wider field of view, and is unaffected by superficial calcifications, making it particularly valuable for evaluating deeper structures.9
MRI is highly sensitive for detecting acute inflammatory edema, fatty replacement, and muscle atrophy. T2-weighted and short tau inversion recovery sequences highlight muscle edema, which appears hyperintense due to increased water content, a hallmark of active muscle inflammation in dermatomyositis. T1-weighted imaging is useful for assessing muscle atrophy, with affected muscle appearing hyperintense, reflecting muscle loss and fatty infiltration. Post-contrast MRI highlights areas of active inflammation, with findings ranging from minimal skin or subcutaneous fat involvement to widespread muscle and myofascial disease.10
US is a valuable tool in assessing disease activity, distinguishing between active and chronic disease, detecting subclinical involvement, and confirming remission.10 Increased muscle echogenicity suggests inflammation and edema, while a heterogeneous echotexture of the muscle reflects varying degrees of inflammation and fibrosis. In early disease, muscles appear swollen and thickened due to active inflammation. In chronic dermatomyositis, the muscle becomes thin and atrophic, reflecting prolonged inflammation and muscle damage.10
While dermatomyositis lacks a curative treatment, extended periods of remission are attainable. With appropriate therapy, disease-related mortality in juvenile dermatomyositis is 2-3%. Glucocorticoids remain the first-line treatment, with prednisone being the most used corticosteroid. Corticosteroid-sparing agents, such as methotrexate or azathioprine, are often introduced to minimize long-term steroid use. In children with severe muscle weakness, high-dose prednisone may be initiated for rapid disease control. However, prolonged glucocorticoid use carries risks, including weight gain, behavioral changes, hypertension, increased infection risk, and impaired bone growth.
Because methotrexate takes approximately 3 months to become effective, it is not the preferred choice in patients requiring immediate disease control. Once clinical improvement is observed, glucocorticoids are gradually tapered to the lowest effective dose to maintain disease control and normalize enzyme levels. Biologic therapies, such as rituximab and anti-tumor necrosis factor agents, are reserved for refractory cases and are not routinely used.6
Conclusion
Juvenile dermatomyositis is a rare autoimmune inflammatory myopathy characterized by progressive proximal muscle weakness and distinctive skin manifestations. Imaging, particularly MRI, plays a crucial role in diagnosis and monitoring, detecting muscle edema, inflammation, and chronic atrophy. Calcinosis is a common complication, particularly in children, and serves as a marker of disease progression. Treatment primarily involves glucocorticoids, with immunosuppressive agents used to minimize long-term steroid exposure. While remission is achievable, ongoing management is necessary to prevent relapses and complications.
References
- Qudsiya Z, Dermatomyositis W. In: StatPearls [Internet]. 2023.
- Tansley S, McHugh N, Wedderburn L. Adult and juvenile dermatomyositis: are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms?. Arthritis Res Ther. 2013;15(2):211. doi:10.1186/ar4198.
- Gara S, Jamil R, Muse M. In: StatPearls [Internet].
- Boyarchuk O, Kuka A, Yuryk I. Clinical and autoantibody phenotypes of juvenile dermatomyositis. Reumatologia. 2022;60(4):281-291. doi:10.5114/reum.2022.119045.
- Kwon K, Lee J, Kim Y. A case report of life-threatening acute dysphagia in dermatomyositis: challenges in diagnosis and treatment. Medicine. 2018;97(17). doi:10.1097/MD.0000000000010508.
- Sudoł-Szopińska I, Jacques T, Gietka P, Cotten A. Imaging in dermatomyositis in adults and children. J Ultrason. 2020;20(80). doi:10.15557/JoU.2020.0007.
- Sanyal S, Atwal S, Mondal D, Garga U. Radiographic patterns of soft tissue calcinosis in juvenile dermatomyositis and its clinical implications. J Clin Diagn Res. 2014;8(12):RD08-RD11. doi:10.7860/JCDR/2014/10787.5321.
- Davuluri S, Duvvuri B, Lood C, Faghihi-Kashani S, Chung L. Calcinosis in dermatomyositis: origins and possible therapeutic avenues. Best Pract Res Clin Rheumatol. 2022;36(2):101768. doi:10.1016/j.berh.2022.101768.
- Spalkit S, Sinha A, Prakash M, Sandhu M. Dermatomyositis: patterns of MRI findings in muscles, fascia and skin of pelvis and thigh. Eur J Radiol. 2021;141:109812. doi:10.1016/j.ejrad.2021.109812.
- Kasapcopur O, Barut K, Avar P. Juvenile dermatomyositis: clinical features, laboratory findings, treatment modalities and disease course (a single center experience). Pediatr Rheumatol. 2014;12(s1):P276. doi:10.1186/1546-0096-12-S1-P276.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Juvenile Dermatomyositis. Journal of Pediatric Case Reports. 2026;1(3). doi:10.37549/JPCR-26-0090.