1 Faculty of Medicine, Setif University, Sétif, Algeria
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 University of Cincinnati College of Medicine, Cincinnati, Ohio
4 Department of Radiology, Cincinnati Children’s Hospital, Cincinnati, Ohio
* Corresponding author: Richard B. Towbin (rtowbin@gmail.com)
Abstract
Leigh syndrome is a rare genetic neurometabolic disorder characterized by the degeneration of the central nervous system. The patient may present with progressive neurological deterioration and include loss of previously acquired motor skills, loss of appetite, vomiting, and irritability. As Leigh syndrome progresses, symptoms may also include a lack of muscle tone, brainstem dysfunction, and episodes of lactic acidosis. Imaging findings of Leigh syndrome consist of bilateral symmetrical hyperintense lesions in the brainstem and basal ganglia.
Keywords
syndrome, genetic, metabolic
Categories
Clinical Summary
A preteen with Leigh syndrome (LS) (due to homozygous mutation in the NDUFS2 gene) presented with worsening balance, diminished hand control, and dysphagia. An MRI of the brain demonstrated multiple new parenchymal lesions. Of note, MR spectroscopy of the brain 5 years prior did not demonstrate an inverted doublet at 1.33 ppm, which indicates an elevated lactate.
Imaging Findings
MRI of the brain demonstrates multifocal areas of abnormal signal intensity and diffusion (Figures 1-3).
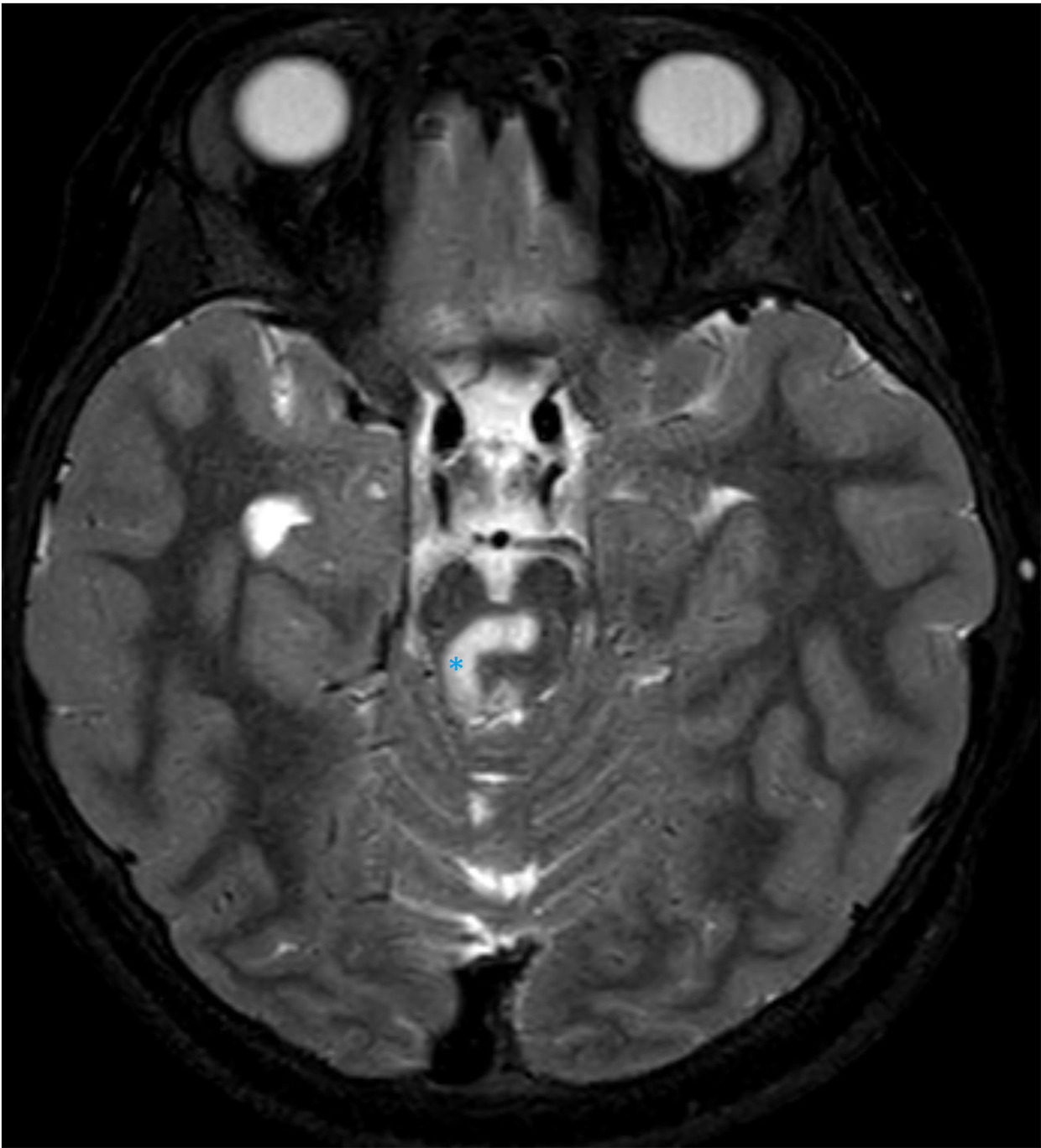
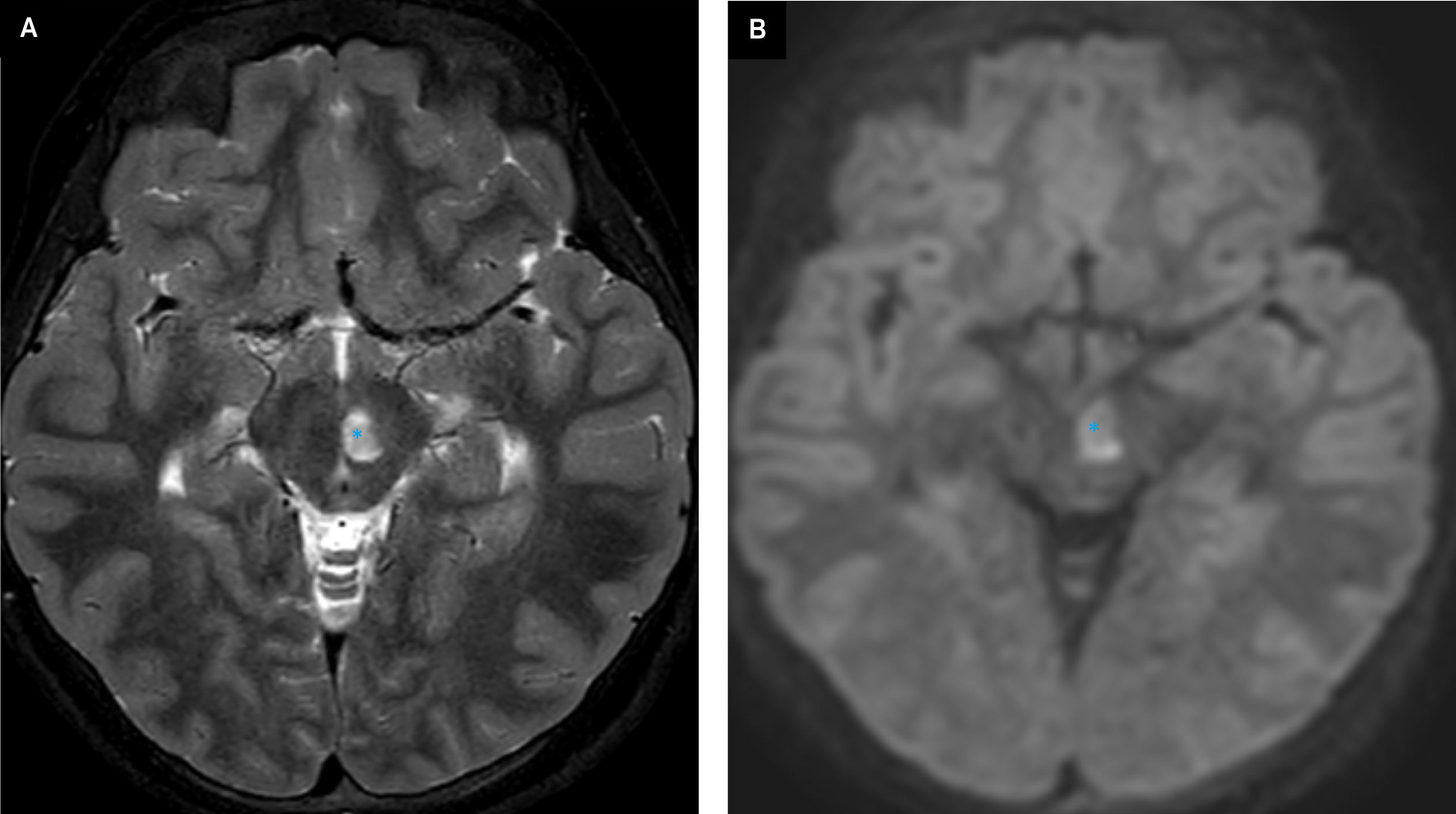
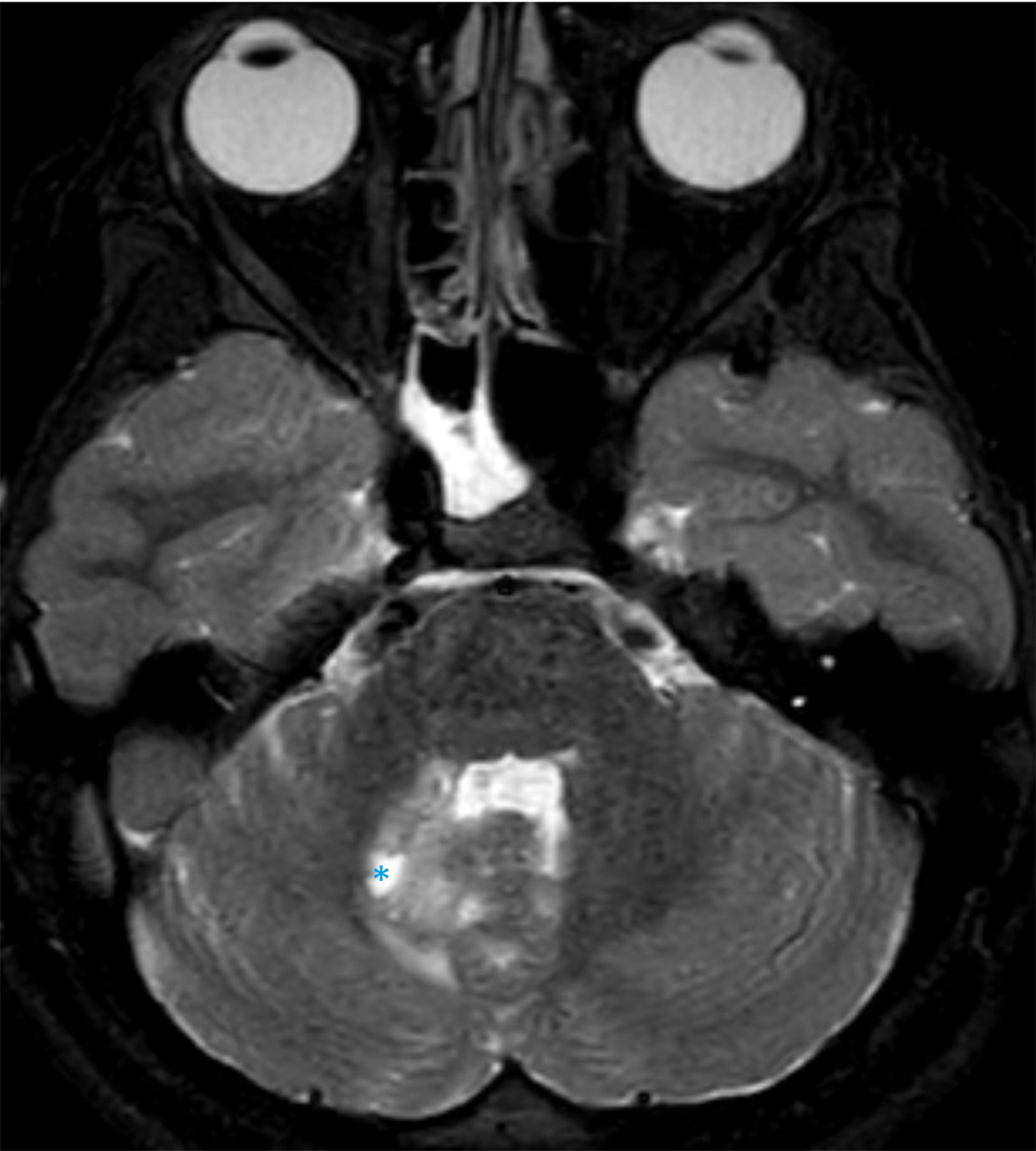
Diagnosis
Leigh syndrome
Differential Diagnosis
Perinatal asphyxia, kernicterus, carbon monoxide or methanol intoxication, thiamine deficiency, Wilson’s disease, and biotin-responsive basal ganglia disease.
Discussion
LS, also referred to as subacute necrotizing encephalopathy, was first described by the British psychiatrist and neuropathologist Denis Archibald Leigh in 1951. 1
It is a rare genetic neurometabolic disorder and childhood’s most common mitochondrial disease. LS is characterized by the degeneration of the central nervous system due to mitochondrial DNA (mtDNA) MT-ATP6 gene or nuclear DNA (nDNA) mutations that affect infants, children, and sometimes adults. 2 LS has an estimated prevalence of 1 per 40,000 live births. Much higher incidences of the condition have been observed in specific populations, for example, Lebanese population, owing to a founder mutation. 3
Most individuals with LS have defects in mitochondrial energy production, such as defects of the pyruvate dehydrogenase complex (PDHC), and deficiency of an enzyme of the mitochondrial respiratory chain complexes I, II, IV, and V. 4,5 In most cases, LS is inherited as an autosomal-recessive trait. However, X-linked recessive and maternal inheritance, due to an mtDNA mutation, are additional modes of transmission.
Although most patients with LS have a mutation in nDNA, about 25% have a mutation in mtDNA. Whether a result of nuclear- or mtDNA-encoded mutation, ultimately it is an impaired oxidative phosphorylation pathway that leads to a critical nadir of cellular energy and subsequent cell death.
The most common causes of nuclear-encoded syndrome are defects in the PDHC or cytochrome c oxidase. The most frequently mutated gene in COX-deficient LS is nuclear-encoded SURF1 gene, which is associated with cognitive sparing and abnormal respiratory chain enzyme activity that was found in most patients with complex 1 (C1) deficiency. NDUFS1 or NDUFS4 is the most common cause of C1 deficiency, and both variants are poor prognostic signs. Other genes have been shown to cause LS such as the mutation of the LRPPRC gene that manifests with mild facial dysmorphism, liver pathology, and a course punctuated by episodes of acute metabolic decompensation. The SCO2 gene mutation can cause hypertrophic cardiomyopathy and spinal muscular atrophy pattern of histopathology in skeletal muscle. 6
mtDNA point mutations have been identified in mtDNA from patients with a variety of disorders; among them, 2 syndromes are more common. The first is MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), which usually presents in children or young adults after normal early development. The second syndrome is MERRF (myoclonus epilepsy with ragged red fibers), characterized by myoclonus, seizures, mitochondrial myopathy, and cerebellar ataxia. 2
The symptoms of LS usually begin between the ages of 3 months and 2 years, 7 but some patients do not exhibit signs and symptoms until several years later. Symptoms are associated with progressive neurological deterioration and include loss of previously acquired motor skills, loss of appetite, vomiting, irritability, seizure activity, hypertrophic cardiomyopathy, chorea, and atrophy of the optic nerve. As LS progresses, symptoms may also include generalized weakness, hypotonia, brainstem dysfunction, and episodes of lactic acidosis, which may lead to impairment of respiratory and kidney function. 8 The prognosis is poor, and death usually occurs in childhood due to respiratory failure.
Diagnosis requires clinical evidence of brainstem and/or basal ganglia dysfunction, 2 intellectual and motor developmental delay, and 3 abnormal energy metabolism indicated by a severe defect in oxidative phosphorylation or PDHC activity.
MRI is the preferred imaging modality. On brain MRI, bilaterally, symmetric findings are seen in the deep gray matter, especially in the basal ganglia and brainstem. The putamen, caudate nucleus, and globus pallidus are the most often involved components of the basal ganglia while the midbrain and medulla are the most frequently involved areas of the brainstem. 9 Hong and colleagues found the basal ganglia and atrophy were the most common findings in 94.5% and 58 % of patients, respectively, followed by brainstem involvement and white matter signal abnormalities in 41% and 38% of patients 10 ; other areas involved include the cerebral peduncles, pons, and medulla.
The prognosis for children with LS is poor. Up to 50% of patients die by age 3 years and about 90% by age 6. The cause of death is usually the result of sepsis, heart failure, neurologic causes, or respiratory failure.
Conclusion
LS is a rare genetic neurometabolic disorder characterized by the degeneration of the central nervous system. The patient may present with progressive neurological deterioration and include loss of previously acquired motor skills, loss of appetite, vomiting, and irritability. As LS progresses, symptoms may also include a lack of muscle tone, brainstem dysfunction, and episodes of lactic acidosis. Imaging findings of LS consist of bilateral symmetrical hyperintense lesions in the brainstem and basal ganglia.
References
- Hirano M. Leigh syndrome. Encyclopedia of Movement Disorders. 2010:125-128. doi:10.1016/B978-0-12-374105-9.00339-7.
- DiMauro S. Mitochondrial diseases. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2004;1658(1-2):80-88. doi:10.1016/j.bbabio.2004.03.014.
- Morin C, Mitchell G, Larochelle J. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am J Hum Genet. 1993;53(2):488-496.
- Ruhoy I, Saneto R. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014;7:221-234. doi:10.2147/TACG.S46176.
- Saneto R, Friedman S, Shaw D. Neuroimaging of mitochondrial disease. Mitochondrion. 2008;8(5-6):396-413. doi:10.1016/j.mito.2008.05.003.
- Lake N, Compton A, Rahman S, Thorburn D. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79(2):190-203. doi:10.1002/ana.24551.
- Sofou K, De Coo I, Isohanni P. A multicenter study on leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9(1). doi:10.1186/1750-1172-9-52.
- Gerards M, Sallevelt S, Smeets H. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. 2016;117(3):300-312. doi:10.1016/j.ymgme.2015.12.004.
- Ardissone A, Bruno C, Diodato D. Clinical, imaging, biochemical and molecular features in Leigh syndrome: a study from the Italian network of mitochondrial diseases. Orphanet J Rare Dis. 2021;16(1):1-12. doi:10.1186/S13023-021-02029-3/TABLES/2.
- Hong C, Na J, Park S, Lee Y. Clinical characteristics of early-onset and late-onset Leigh syndrome. Front Neurol. 2020;11. doi:10.3389/fneur.2020.00267.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Leigh Syndrome. Applied Radiology. 2025. doi:10.37549/JPCR-25-0010.