Pulmonary Interstitial Glycogenosis
Journal of Pediatric Case Reports — Vol. 1 , Issue 3
Published: April 1, 2026
1 Kansas City University College of Osteopathic Medicine, Kansas City, Missouri
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 Department of Radiology, Cincinnati Children’s Hospital, University of Cincinnati College of Medicine, Cincinnati, Ohio
* Corresponding author: Richard B. Towbin (rtowbin@gmail.com)
Abstract
Pulmonary interstitial glycogenosis (PIG) is a rare interstitial lung disease that presents in neonates with rapidly progressive respiratory distress. Because there are no specific clinical, radiologic, or genetic markers for the disease, diagnosis relies on lung biopsy. Prognosis depends on disease context: isolated PIG is typically associated with favorable outcomes, whereas cases with concurrent pulmonary or cardiovascular abnormalities may have a more complicated clinical course. Treatment generally includes corticosteroids and supportive care, tailored to the severity of respiratory symptoms and any underlying conditions.
Keywords
thorax, lung, congenital, cause unknown
Categories
Case Summary
An infant born at 34 weeks started grunting in the neonatal intensive care unit and ultimately required continuous positive airway pressure ventilation, exogenous surfactant, and intubation. After extubating, the patient required oxygen.
Imaging Findings
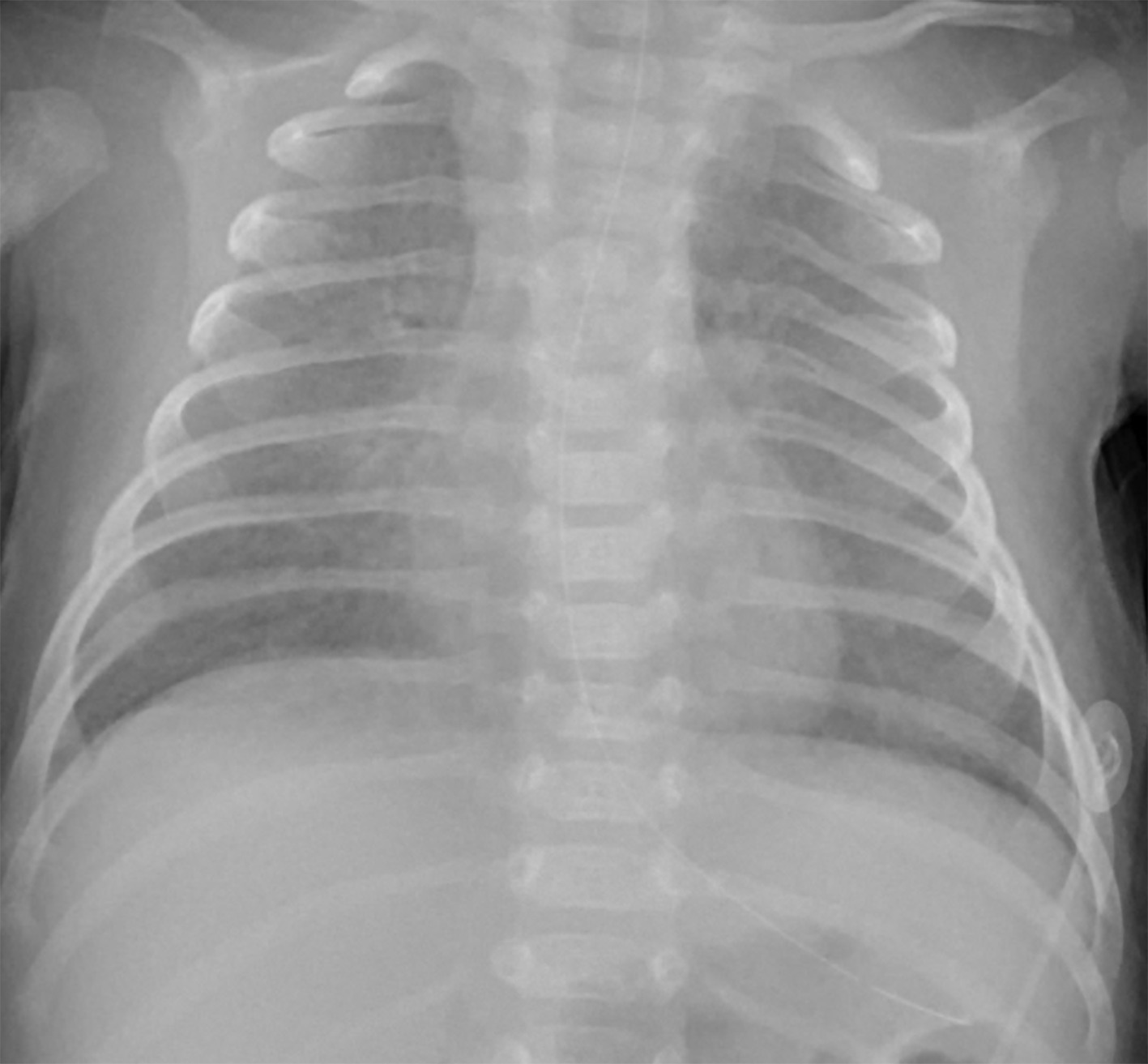
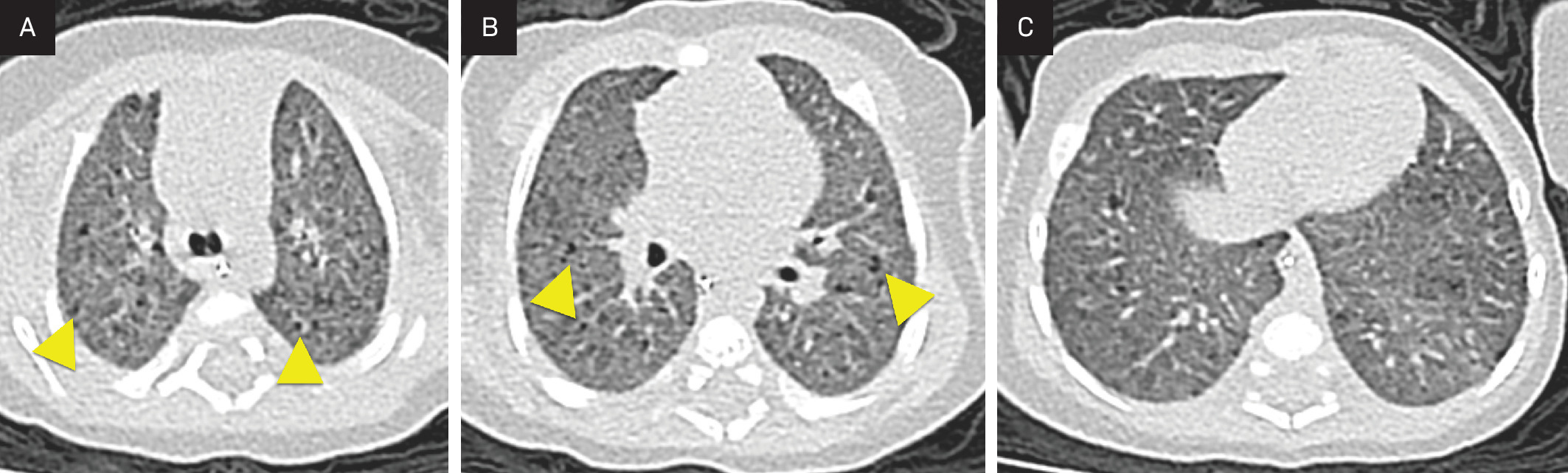
Radiograph (Figure 1) and chest CT (Figure 2) were obtained at 6 weeks of age. Both show diffuse ground-glass opacity and small cystic spaces in the posterior aspect of the lungs.
Diagnosis
Pulmonary interstitial glycogenosis (PIG).
Differential diagnosis includes bronchopulmonary dysplasia (chronic lung disease of prematurity), neuroendocrine hyperplasia of infancy, surfactant dysfunction disorders, and glycogen storage diseases.
Discussion
PIG, previously termed cellular interstitial pneumonitis or histiocytoid pneumonia, is a rare form of neonatal interstitial lung disease that typically affects infants younger than 6 months of age.1 The exact cause remains unknown, but PIG is thought to be a congenital abnormality resulting from abnormal alveolar growth or impaired lung remodeling, leading to restricted differentiation of pulmonary interstitial fibroblasts.2,3 This process results in the accumulation of glycogen within the interstitial cells of the lung.
PIG remains poorly understood, with most of the literature limited to case reports. Its reported incidence ranges from 0.1 to 16 cases per 100,000 children per year, with a slight male predominance.4 PIG occurs in both term and preterm infants and is frequently associated with other congenital lung abnormalities and cardiovascular disease, including pulmonary hypertension, structural heart defects, alveolar simplification, and extrapulmonary conditions such as Noonan syndrome, inborn errors of metabolism, and Trisomy 21.1,2,5,6 PIG may also present as an isolated finding, in which case the clinical course is often favorable.2
Clinically, neonates and infants with PIG present with rapidly progressive respiratory distress, tachypnea, and hypoxemia in the absence of infection.7,8 After an initial period of stability, respiratory function may deteriorate, requiring varying levels of support. When PIG is suspected, the gold standard for diagnosis is lung biopsy, which reveals expansion of the interstitium by spindle-shaped mesenchymal cells with pale cytoplasm containing glycogen.2,3
High-resolution chest CT in PIG typically demonstrates features of interstitial lung disease, though no uniform imaging pattern has been established. The most common findings include diffuse ground-glass opacities and cystic lucencies, which are most pronounced in the posterior lungs.5,9 These cystic lucencies are not true cysts but instead correspond to areas of alveolar simplification.2 Less frequent imaging findings in PIG include interlobular septal thickening and architectural distortion. The extent of imaging findings does not correlate with the degree of lung growth abnormality or clinical outcome.2
There is no established consensus for the treatment of PIG. Systemic corticosteroids, typically administered as monthly pulses, are commonly used with anecdotal reports of clinical improvement.5,10 Although PIG lacks active inflammation, corticosteroids are thought to promote lung maturation, potentially by accelerating apoptosis of lipofibroblasts.2,5 Additional supportive therapies include oxygen supplementation and pulmonary rehabilitation in milder cases, while severe cases may require extracorporeal membrane oxygenation or even lung transplantation.1,5
Overall, prognosis depends largely on the extent of lung development and the presence of associated comorbidities. Patients with isolated PIG tend to experience significant clinical improvement and a favorable outcome, whereas those with underlying abnormalities have a more variable prognosis.1,5
Conclusion
PIG is a rare interstitial lung disease that presents in neonates with rapidly progressive respiratory distress. Because there are no specific clinical, radiologic, or genetic markers for the disease, diagnosis relies on lung biopsy. Prognosis depends on disease context: isolated PIG is typically associated with favorable outcomes, whereas cases with concurrent pulmonary or cardiovascular abnormalities may have a more complicated clinical course. Treatment generally includes corticosteroids and supportive care, tailored to the severity of respiratory symptoms and any underlying conditions.
References
- Deutsch G, Cortes-Santiago N. In: Pulmonary Pathology. 2025:62-82. doi:10.1016/B978-0-323-93548-7.00005-2.
- Sardón O, Torrent-Vernetta A, Rovira-Amigo S. Isolated pulmonary interstitial glycogenosis associated with alveolar growth abnormalities: a long-term follow-up study. Pediatr Pulmonol. 2019;54(6):837-846. doi:10.1002/ppul.24324.
- Deutsch G, Young L. Lipofibroblast phenotype in pulmonary interstitial glycogenosis. Am J Respir Crit Care Med. 2016;193(6):694-696. doi:10.1164/rccm.201509-1809LE.
- Das S, Langston C, Fan L. Interstitial lung disease in children. Curr Opin Pediatr. 2011;23(3):325-331. doi:10.1097/MOP.0b013e3283464a37.
- Liptzin D, Baker C, Darst J. Pulmonary interstitial glycogenosis: diagnostic evaluation and clinical course. Pediatr Pulmonol. 2018;53(12):1651-1658. doi:10.1002/ppul.24123.
- Cutz E, Chami R, Dell S, Langer J, Manson D. Pulmonary interstitial glycogenosis associated with a spectrum of neonatal pulmonary disorders. Hum Pathol. 2017;68:154-165. doi:10.1016/j.humpath.2017.06.026.
- Griese M, Seidl E. Persistent tachypnea of infancy, neuroendocrine cell hyperplasia of infancy, and pulmonary interstitial glycogenosis: “A3-specific conditions of undefined etiology”. Pediatr Pulmonol. 2024;59(10):2702-2707. doi:10.1002/ppul.27102.
- Seidl E, Carlens J, Reu S. Pulmonary interstitial glycogenosis - a systematic analysis of new cases. Respir Med. 2018;140:11-20. doi:10.1016/j.rmed.2018.05.009.
- Weinman J, White C, Liptzin D. High-resolution CT findings of pulmonary interstitial glycogenosis. Pediatr Radiol. 2018;48(8):1066-1072. doi:10.1007/s00247-018-4138-4.
- Hamberger E, Yu Y, Choi H. Pulmonary interstitial glycogenosis in two neonates: early recognition and use of corticosteroids. Respir Med Case Rep. 2024;48. doi:10.1016/j.rmcr.2024.101990.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Pulmonary Interstitial Glycogenosis. Journal of Pediatric Case Reports. 2026;1(3). doi:10.37549/JPCR-26-0089.