Von Hippel-Lindau Syndrome
Applied Radiology — Vol. 1 , Issue 1 , pp. 1 -3
Published: November 1, 2025
1 Banner University Hospital, Phoenix, Arizona
2 Department of Radiology, Phoenix Children’s Hospital, Phoenix, Arizona
3 Department of Radiology, University of Cincinnati, Cincinnati, Ohio
4 Department of Radiology, Cincinnati Children’s Hospital, Cincinnati, Ohio
* Corresponding author: Richard B. Towbin (rtowbin@gmail.com)
Abstract
VHL is a rare tumor predisposition syndrome characterized by tumors such as hemangioblastoma, renal cell carcinoma, and pheochromocytoma. Although patients are often diagnosed within the 2nd to 3rd decade of life, patients may present with tumors during childhood. Patients are diagnosed based on family history, clinical findings, and genetic testing. Long-term follow-up and screening is necessary due to the high likelihood of tumors throughout the body.
Keywords
syndrome, genetic, Brain, Neoplasm, Eye
Categories
Case Summary
A pre-teen girl presented with worsening headaches, disequilibrium and vomiting. Her family history was significant for unspecified cancers in her father and maternal grandmother. Ophthalmic examination showed multiple retinal hemangioblastoma. Over time, the patient had multiple craniotomies for brain tumor resections. Additionally, she had multiple treatment approaches for her right eye, including cryotherapies, laser therapies, surgeries, and ultimate enucleation.
Imaging Findings
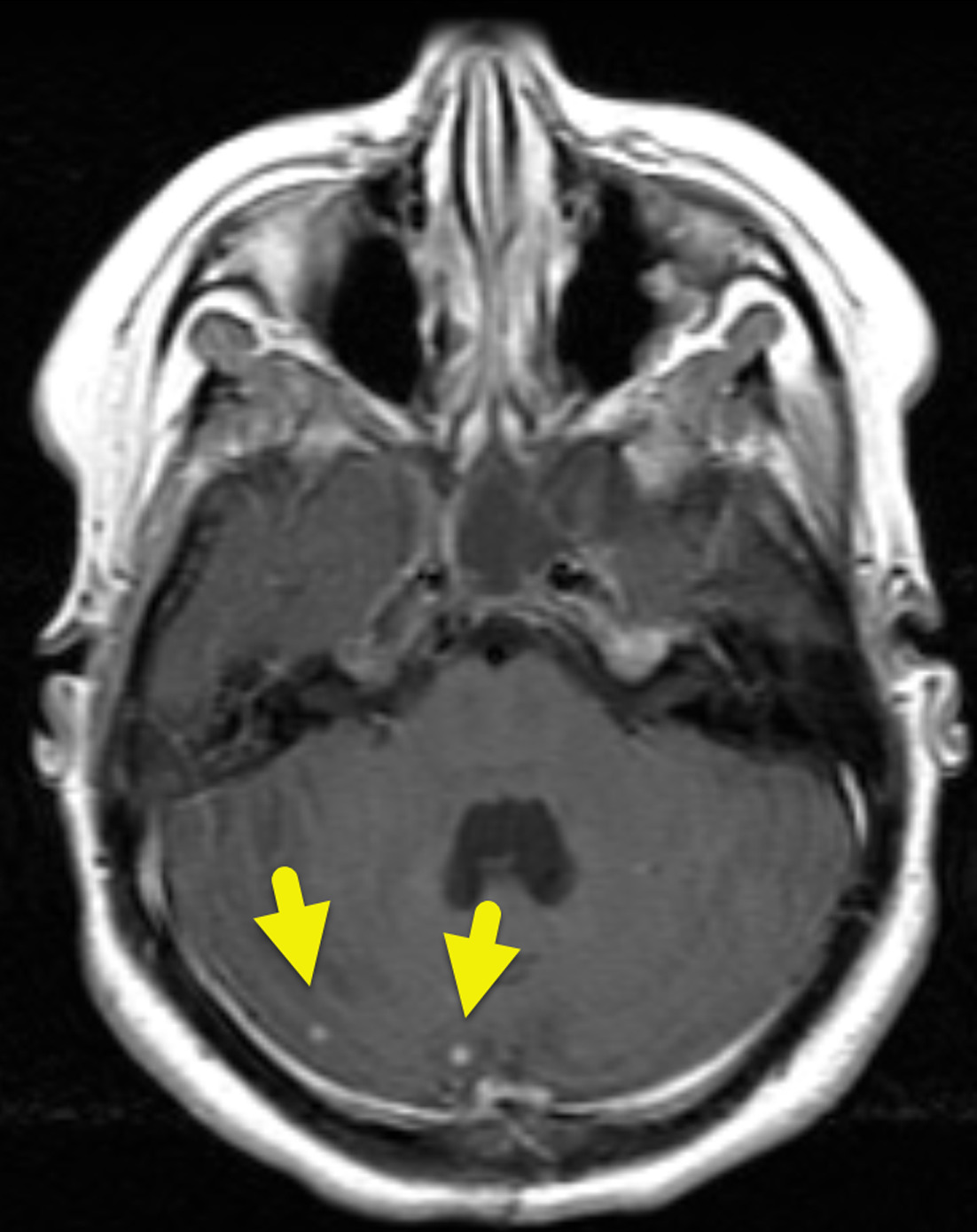
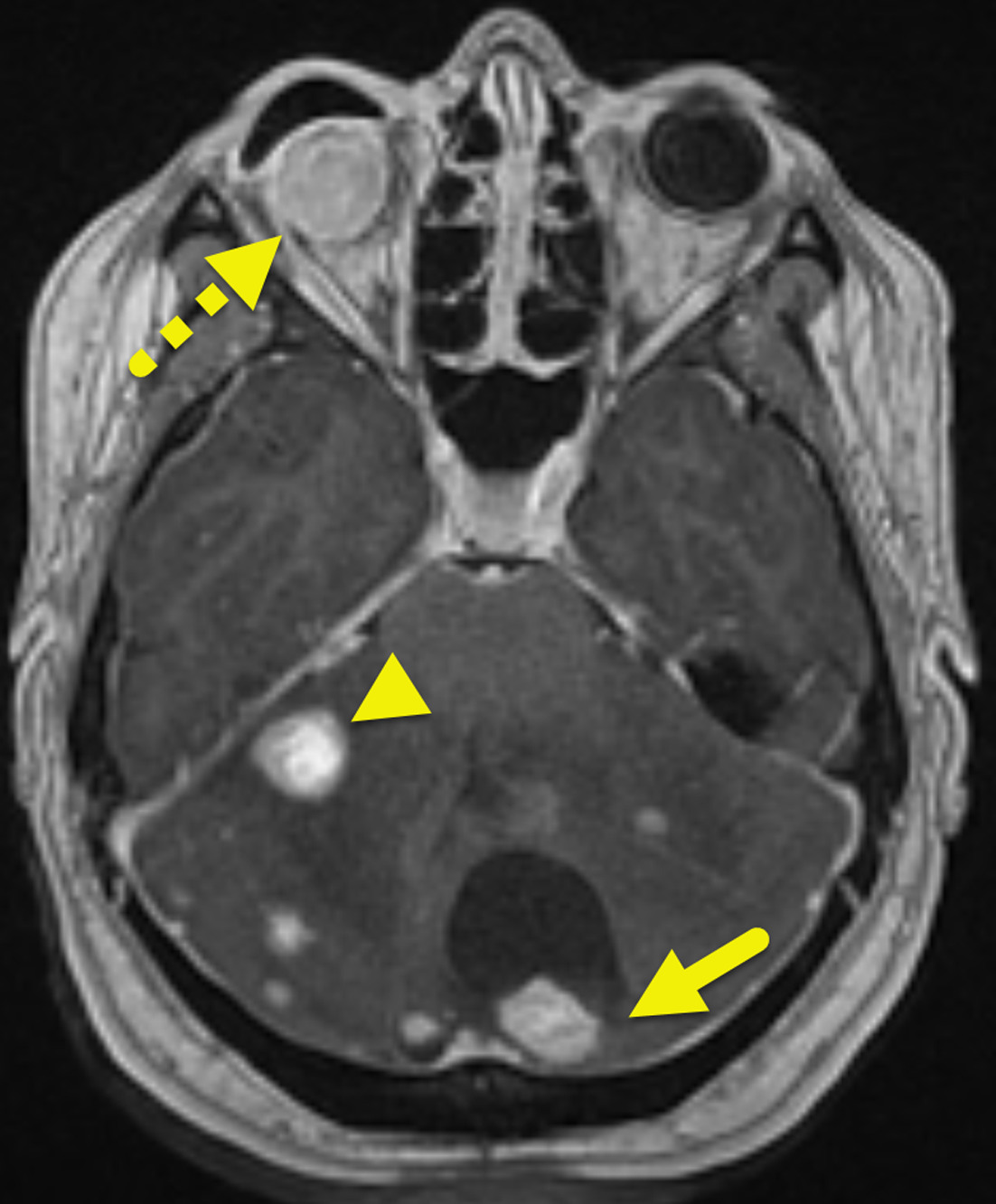
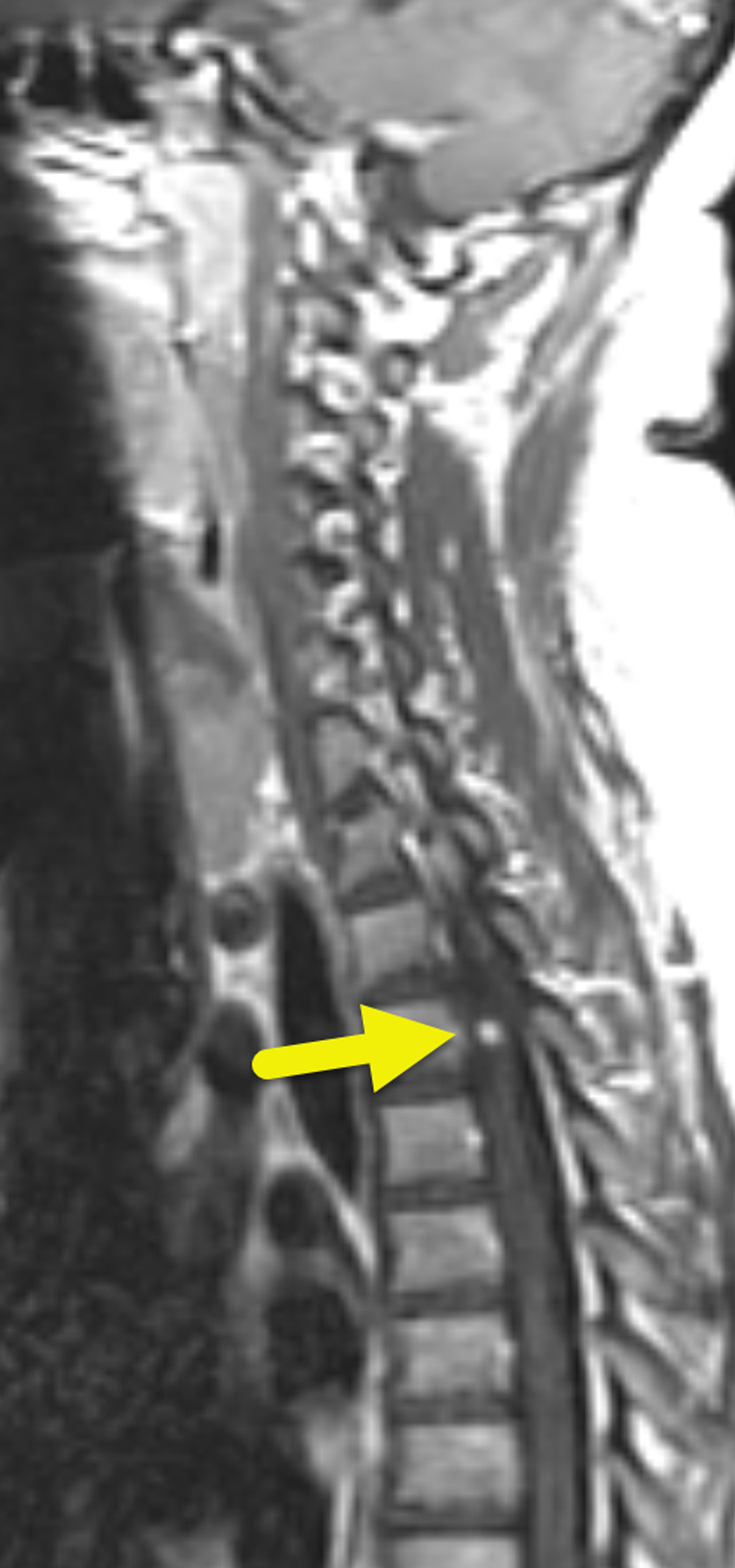
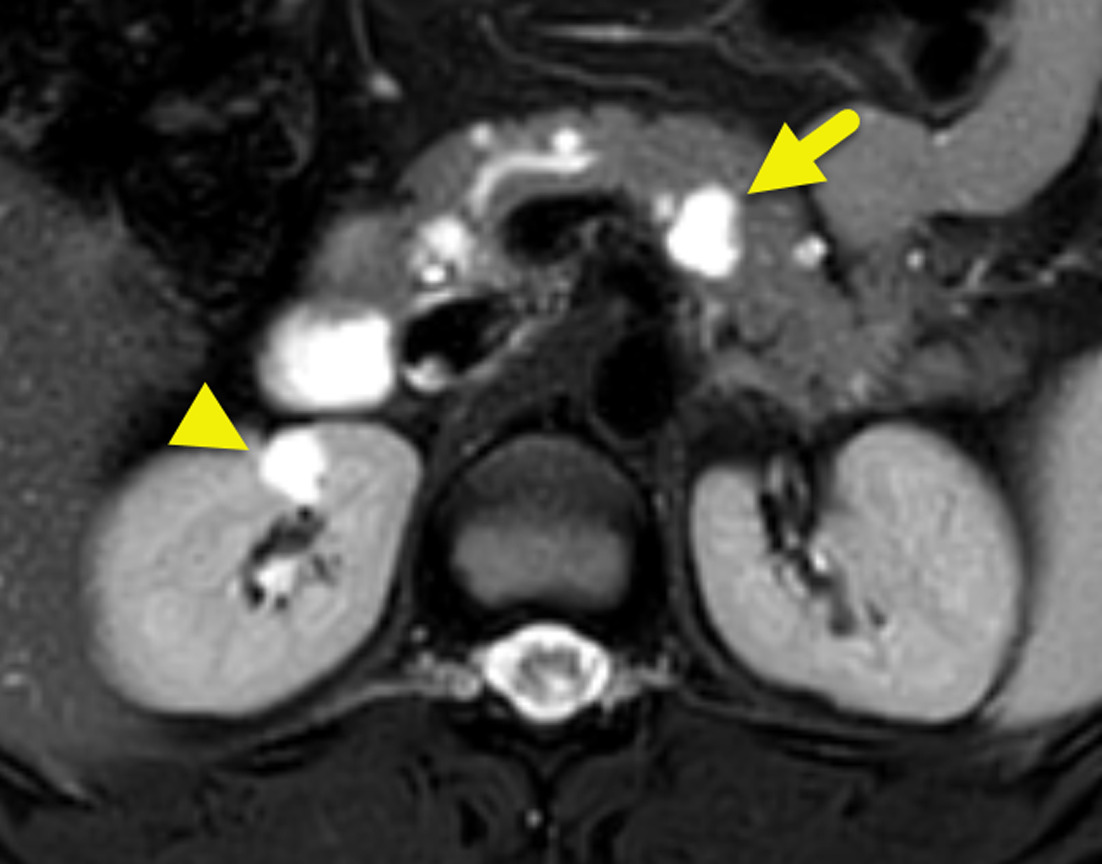
Initial MRI showed multiple small enhancing foci within the cerebellum (Figure 1). These increased in size and number over time (Figure 2). Small enhancing foci were also present in the spinal cord (Figure 3). Abdominal MRI showed multiple pancreatic and renal cysts (Figure 4).
Diagnosis
Multiple cerebellar, spinal cord, and retinal hemangioblastomas in a patient with von Hippel-Lindau (VHL) disease.
Discussion
VHL disease is an autosomal-dominant multisystemic tumor predisposition syndrome characterized by the presence of tumors such as hemangioblastomas and multiple cysts. It occurs because of a mutation of the VHL tumor suppressor gene located on chromosome 3. The mutation leads to dysregulation in the cellular response to hypoxia. 1
VHL occurs in 1 in 36,000 live births. About 80% of patients have an affected parent, with the remaining developing VHL via sporadic mutation. 2 Symptomatic patients present on average at 26 years of age. 2 However, only 50% of patients are symptomatic at the time of presentation. 3 When symptoms are present, they depend on the type of tumor affecting a patient and its location. 4 For example, patients with cerebellar hemangioblastoma may present with headache, vomiting, or ataxia while patients with pheochromocytoma may present with episodic hypertension or flushing. 4
Diagnosis of VHL is made with the combination of clinical symptoms, family history, or genetic testing. In patients with a family history of VHL, diagnosis is made via genetic testing or with the presence of any one of the following: retinal, cerebellar, or spinal hemangioblastoma; renal cell carcinoma; pheochromocytoma or paraganglioma; or multiple renal and pancreatic cysts. If a family history of VHL is not present, a clinical diagnosis requires the presence of 2 or more of the following: 2 or more retinal or central nervous system (CNS) hemangioblastomas or a single hemangioblastoma in association with a visceral manifestation (e.g., multiple kidney or pancreatic cysts); renal cell carcinoma, pheochromocytoma, or paraganglioma; or endolymphatic sac tumors, papillary cystadenomas of the epididymis or broad ligament, or neuroendocrine tumors (NETs) of the pancreas.
Because each of these tumors is rare in children, the presence of any should be an indication to perform genetic testing in the test the patient and their family members. 5 In patients with a diagnosis of VHL, routine screening is recommended. Screening includes annual neurological and ophthalmologic examination, annual blood pressure measurement, annual (starting at 5 years of age) plasma or urine metanephrine levels, and every 2-3 year audiology examination. 5,6 In addition, the following imaging screening tests are recommended: MRI of the brain and total spine every 2 years (starting at 16 years of age), annual abdominal US (starting at 8 years of age), and abdominal MRI every 2 years (starting at 16 years of age).
Patients with VHL may have a variety of manifestations. Hemangioblastomas are the characteristic tumor. They occur within the CNS or retina. CNS hemangioblastomas affect 60-80% of patients with VHL and can occur in the brain (80%) or spinal cord (20%). When hemangioblastomas affect the brain, they are most common in the cerebellum (Figures 1, 2). In the spine, they are most common in the cervical or thoracic cord (Figure 3). On imaging, these tumors appear as a cyst and enhancing mural nodule. 7,8 The size of the cystic component is variable, and small tumors can appear entirely solid.
Retinal hemangioblastomas occur in 70% of patients with VHL. They are identified via ophthalmologic examination. The tumors have a characteristic appearance with arteries and veins and optic disc edema. Although the role of imaging is limited, CT and MRI may show nodular retinal lesions with enhancement, with or without retinal detachment. 7,8
Endolymphatic sac tumors are rare tumors arising from the vestibular aqueduct. The tumor occurs in up to 15% of patients with VHL. Patients commonly present with hearing loss and tinnitus. Both CT and MRI demonstrate an intensely enhancing tumor within the petrous bone. The tumor erodes the bone, giving it a moth-eaten appearance on CT. It is heterogeneous on MRI with foci of hyperintense T1 signal. 8
In the abdomen, several different manifestations may be present. Cysts may develop in the pancreas, liver, kidneys, or genitourinary tract. Tumors can also occur and include pheochromocytomas or paragangliomas, renal cell carcinoma, pancreatic serous cystadenoma, and pancreatic NETs. 9
Renal manifestations, such as renal cell carcinomas or renal cysts, are common and are the most common cause of patient demise in up to 75% of cases. US can differentiate between cystic and solid renal masses. If a solid mass is present, CT or MRI should be performed to evaluate for renal cell carcinoma. Renal cell carcinoma occurs in up to 70% of patients with VHL by 60 years of age and is usually the clear cell variant. On CT, clear cell renal carcinoma appears as a heterogeneous, enhancing, exophytic solid mass. On MRI, the tumor has a heterogeneous appearance on T1-weighted images and is hyperintense on T2-weighted images. 7
Pancreatic manifestations, such as pancreatic cysts or pancreatic NETs, may be seen in 42% of patients with VHL. Cysts are considerably more common than NET. The cysts have a simple appearance and may be numerous (Figure 4). NETs are uncommon, only occurring in 5-17% of patients. When present, they are typically non-functioning and are slow growing. On CT, NETs are hypervascular and may enhance avidly on the arterial phase of imaging. Larger tumors enhance more heterogeneously. On MRI, a NET is hypointense to the pancreas on T1-weighted images and hyperintense on T2-weighted images. The enhancement pattern is like CT. 19F fluorodeoxyglucose (FDG)-PET of pancreatic NETs has shown that a higher maximum standardized uptake value (SUVmax) correlates with tumor size on CT, which may indicate pancreatic lesions with higher malignant/metastatic potential. 10 68Ga-DOTATATE PET/CT is a more specific method to identify the tumor and potential metastases.
Pheochromocytomas and paragangliomas can occur in patients with VHL. Pheochromocytoma is more common, affecting 30% of patients (compared with 15% for paragangliomas). Patients with pheochromocytoma may present with hypertension and flushing while patients with paraganglioma are asymptomatic. On MRI, the tumors are T2-hyperintense. They enhance avidly on both CT and MRI. The tumors also display uptake of radiopharmaceutical on 123I-MIBG scintigraphy, 19F FDG-PET/CT and 68Ga-DOTATATE PET/CT. 7
Reproductive organ manifestations, such as epididymal or broad ligament papillary cystadenomas, may develop in 25-60% of patients with VHL. US demonstrates a cystic mass with a variably solid component and color Doppler flow. 7
The treatment of VHL is multidisciplinary and personalized depending on symptoms and tumor burden. Key indications for neurosurgical hemangioblastoma removal are critically large tumor size, rapid tumor growth, and development of neurological symptoms. Retinal hemangioblastomas are typically treated with laser therapy if small, and with cryocoagulation if large. Enucleation may be necessary in patients with severe pain and blindness. Endolymphatic sac tumors are resected, if possible. Asymptomatic pancreatic NETs have a lower risk of metastasis than sporadic NET and can be monitored via imaging. Pheochromocytomas are treated with surgical excision. For VHL-associated renal cell carcinoma, nephron-sparing surgery is performed for tumors >3 cm, if possible, due to the potential for additional renal tumors. Ablative procedures may be offered to patients with renal tumors <3 cm. 2
Conclusion
VHL is a rare tumor predisposition syndrome characterized by tumors such as hemangioblastoma, renal cell carcinoma, and pheochromocytoma. Although patients are often diagnosed within the 2nd to 3rd decade of life, patients may present with tumors during childhood. Patients are diagnosed based on family history, clinical findings, and genetic testing. Long-term follow-up and screening are necessary due to the high likelihood of tumors throughout the body.
References
- Shulman M, Shi R, Zhang Q. Von Hippel-Lindau tumor suppressor pathways & corresponding therapeutics in kidney cancer. J Genet Genomics. 2021;48(7):552-559. doi:10.1016/j.jgg.2021.05.016.
- Varshney N, Kebede A, Owusu-Dapaah H. A review of von Hippel-Lindau syndrome. J Kidney Cancer VHL. 2017;4(3):20-29. doi:10.15586/jkcvhl.2017.88.
- Mikhail M, Singh A. StatPearls [Internet]. 2022.
- Leeuwaarde R R, Ahmad S, Nesselrooij B, et al. GeneReviews® [Internet]. 2000:1993-2024.
- Ben-Skowronek I, Kozaczuk S. Von Hippel-Lindau syndrome. Horm Res Paediatr. 2015;84(3):145-152. doi:10.1159/000431323.
- Chittiboina P, Lonser R. Von Hippel-Lindau disease. Handb Clin Neurol. 2015;132:139-156. doi:10.1016/B978-0-444-62702-5.00010-X.
- Ganeshan D, Menias C, Pickhardt P. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-of-the-art review. Radiographics. 2018;38(3):849-866. doi:10.1148/rg.2018170156.
- Mourão J, Borella L, Duarte J. Imaging manifestations of von Hippel-Lindau disease: an illustrated guide focusing on the central nervous system. Radiol Bras. 2022;55(3):188-192. doi:10.1590/0100-3984.2021.0080-en.
- Maher E, Neumann H, Richard S. Von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617-623. doi:10.1038/ejhg.2010.175.
- Sadowski S, Weisbrod A, Ellis R. Prospective evaluation of the clinical utility of 18-fluorodeoxyglucose PET CT scanning in patients with von Hippel-Lindau-associated pancreatic lesions. J Am Coll Surg. 2014;218(5):997-1003. doi:10.1016/j.jamcollsurg.2014.01.004.
Disclosures
The authors have no conflicts of interest to disclose. None of the authors received outside funding for the production of this original manuscript and no part of this article has been previously published elsewhere.
Citation
. Von Hippel-Lindau Syndrome. Applied Radiology. 2025;1(1):1-3. doi:10.37549/JPCR-25-0007.